NPM-ALK Is a Key Regulator of the Oncoprotein FOXM1 in ALK-Positive Anaplastic Large Cell Lymphoma

 ,
,  ,
,  and
and 
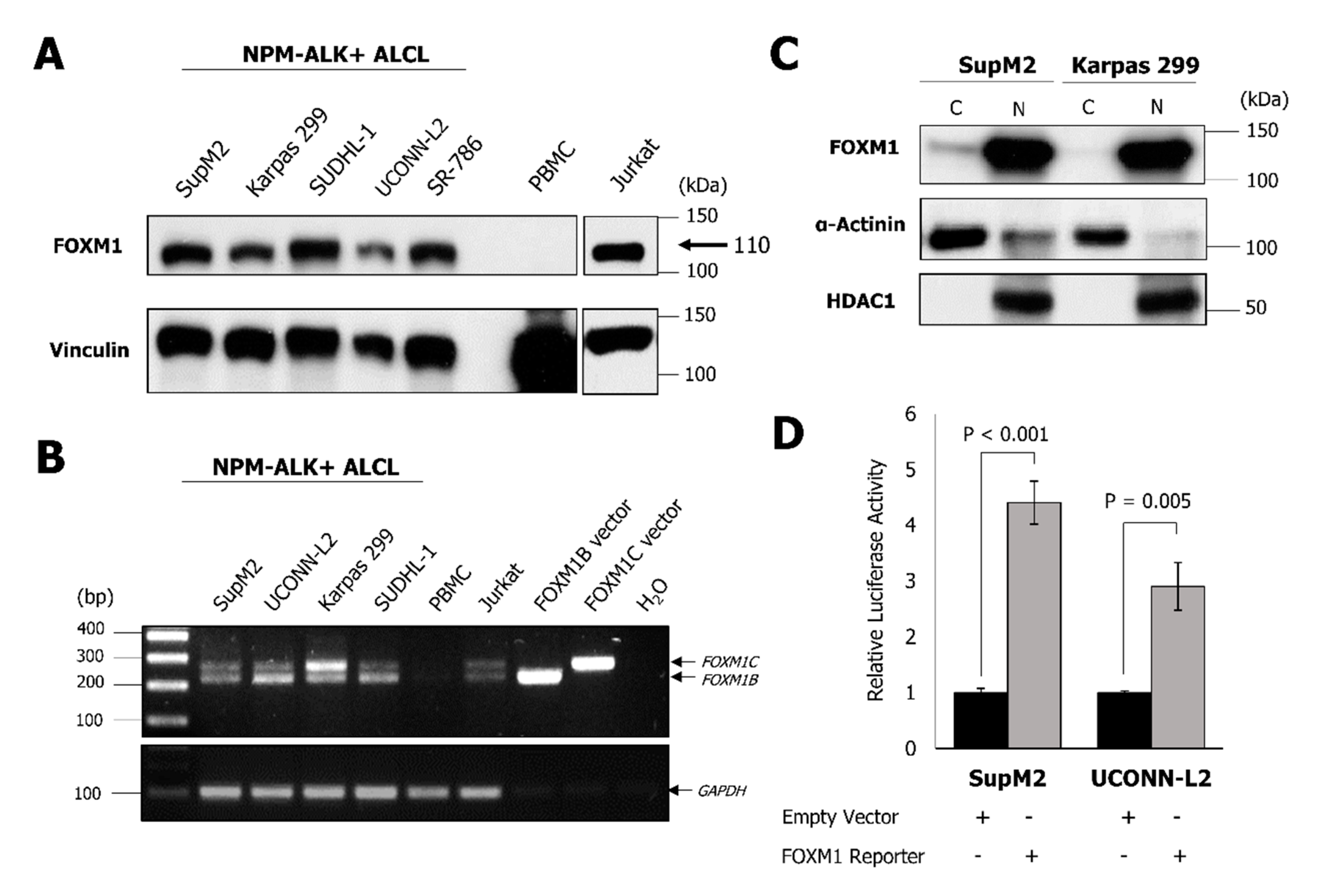{kind=link}
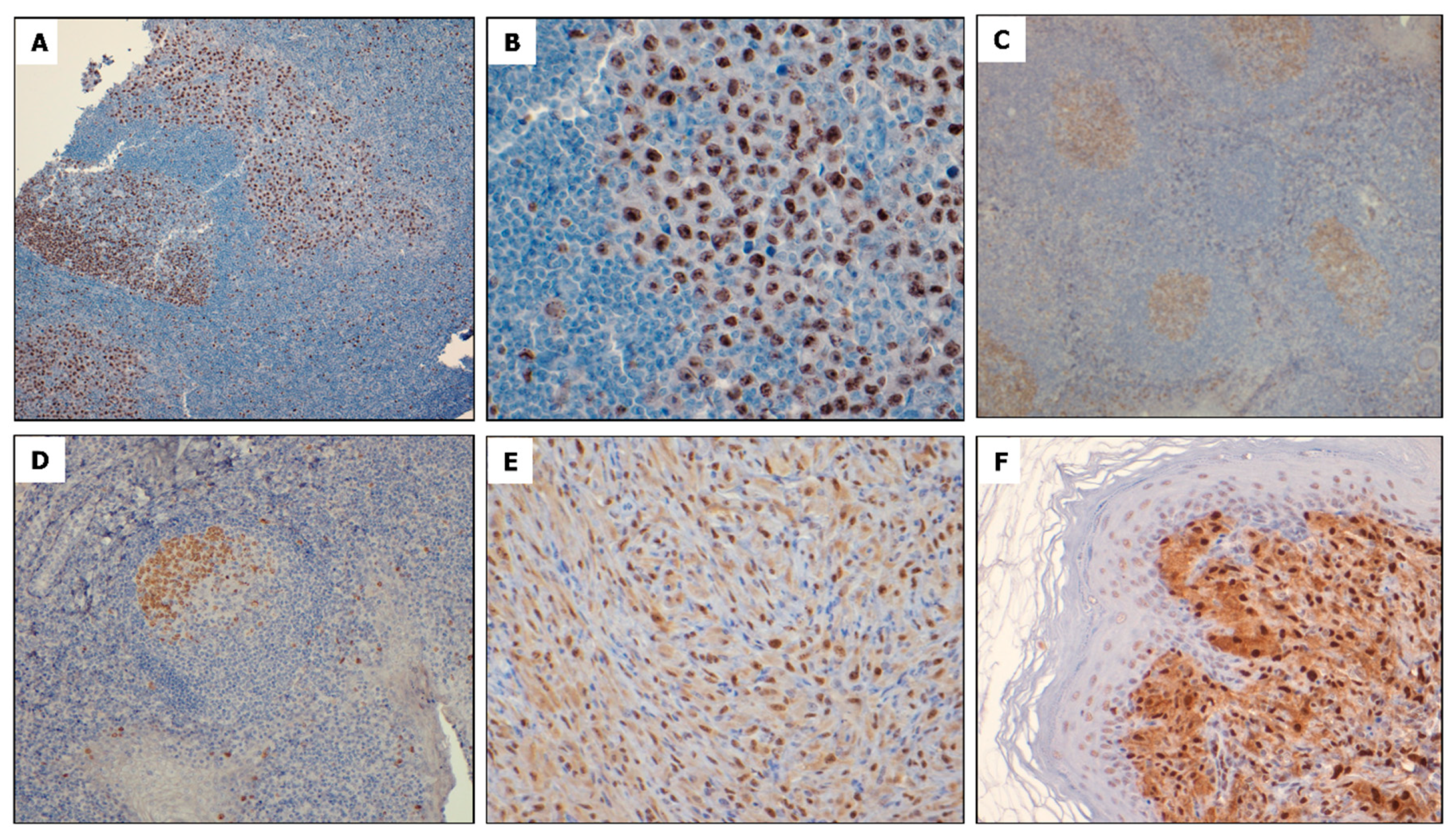{kind=link}
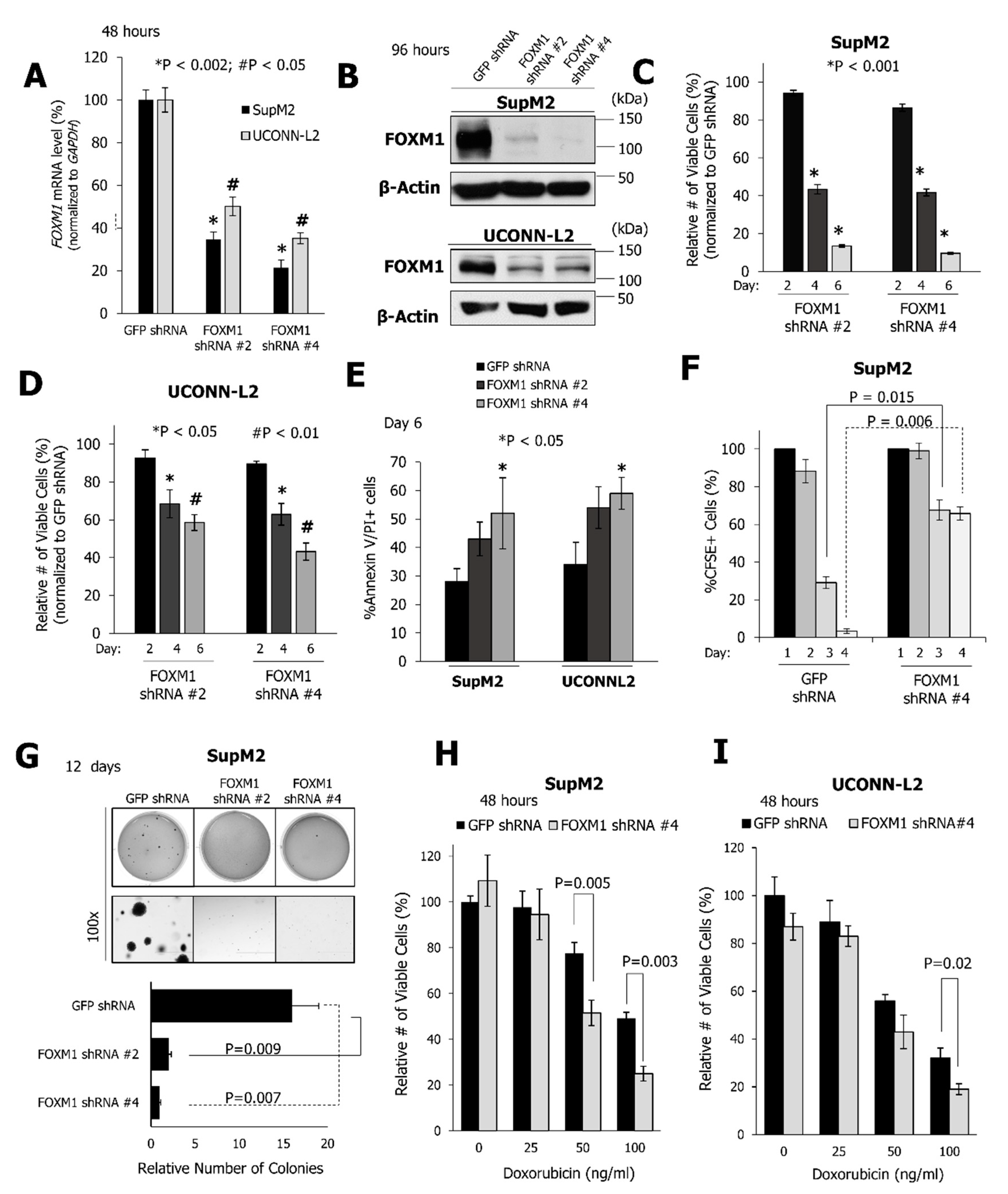{kind=link}
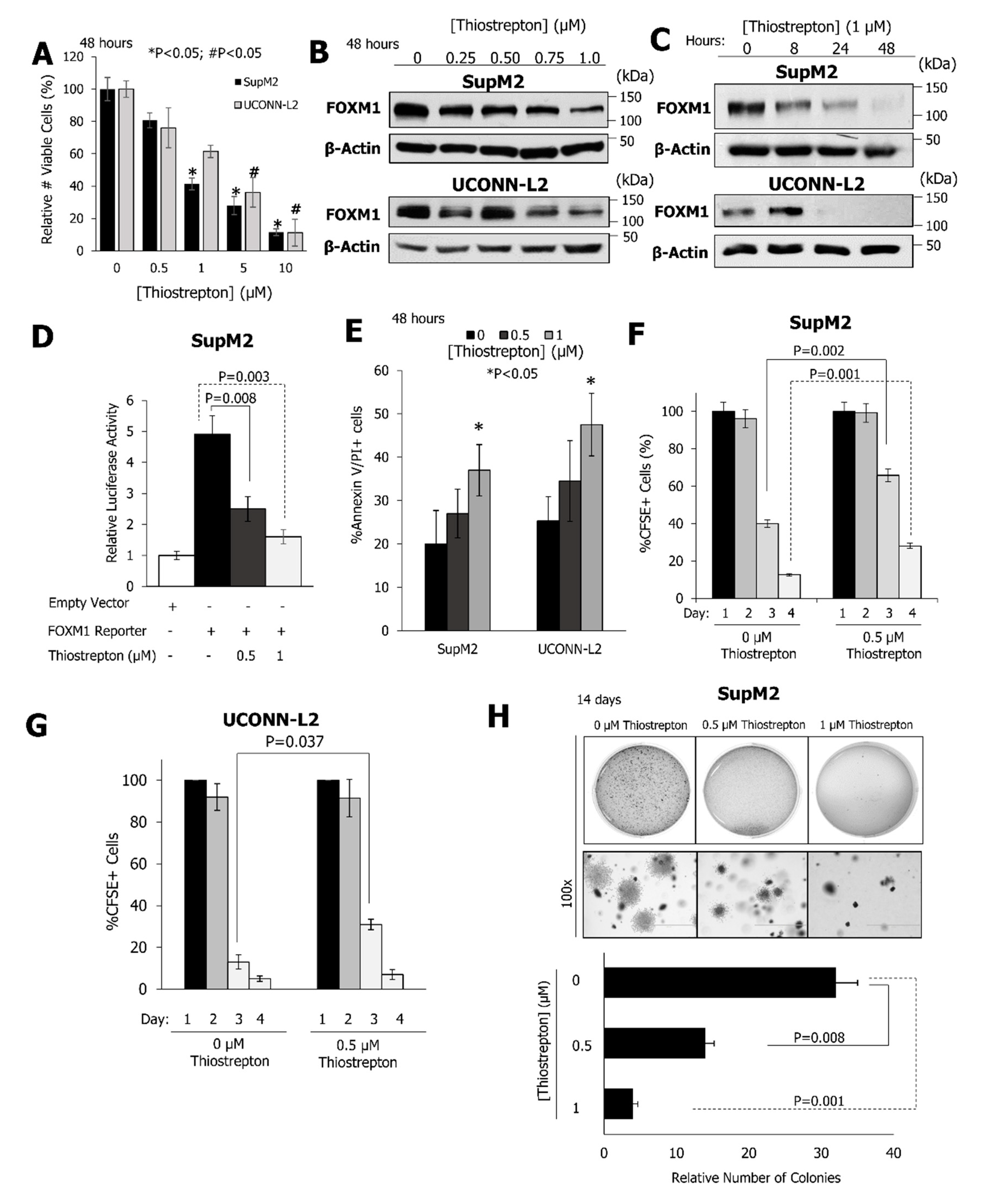{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. NPM-ALK + ALCL Cells Express Elevated FOXM1
2.2. FOXM1 Downregulation Inhibits Cell Growth and the Clonogenicity of NPM-ALK + ALCL Cell Lines
2.3. Pharmacological Inhibition of FOXM1 Inhibits the Growth and Clonogenicity of NPM-ALK + ALCL Cell Lines
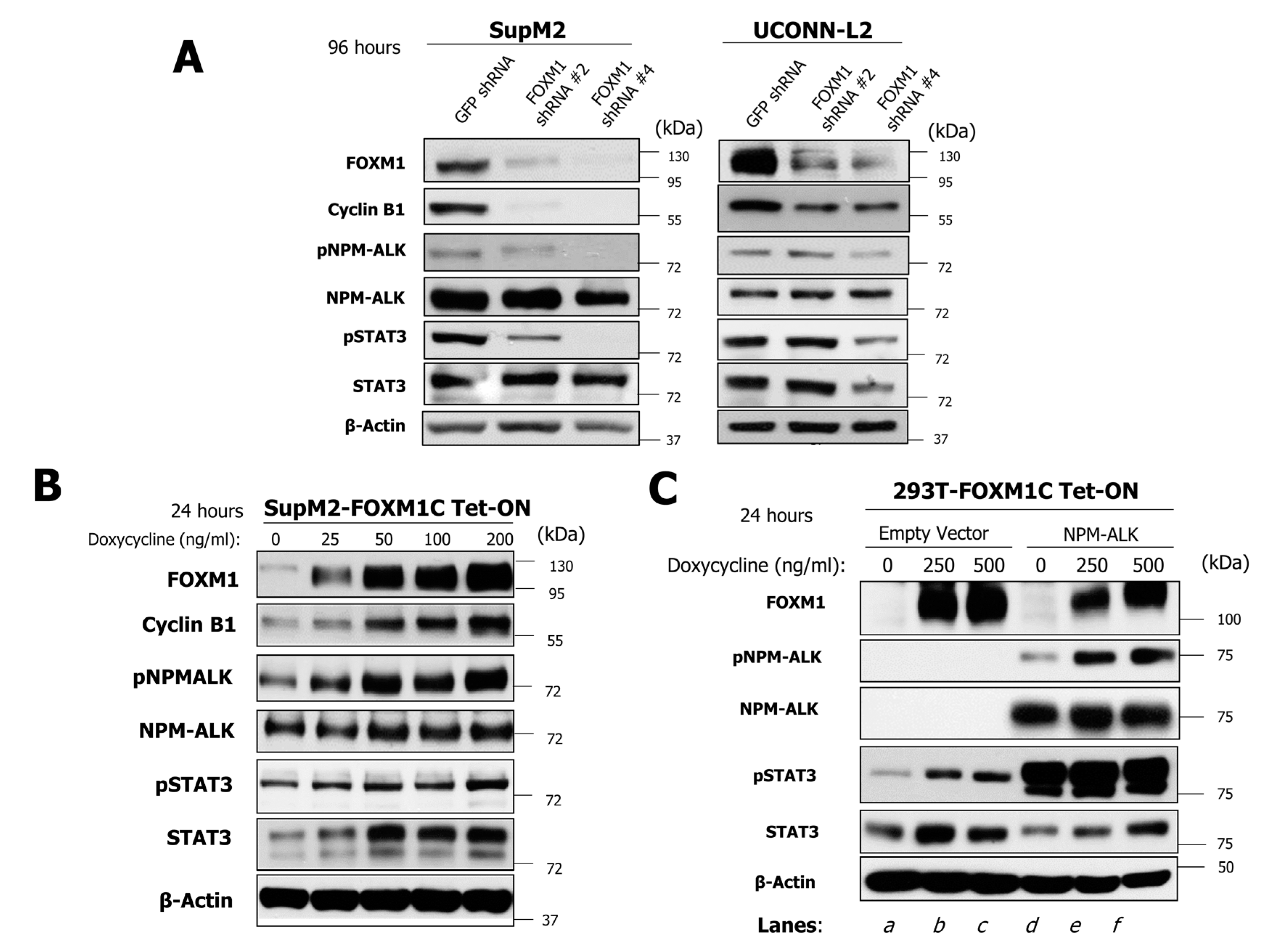
2.4. FOXM1 Induces Phosphorylation of NPM-ALK and STAT3 in NPM-ALK + ALCL
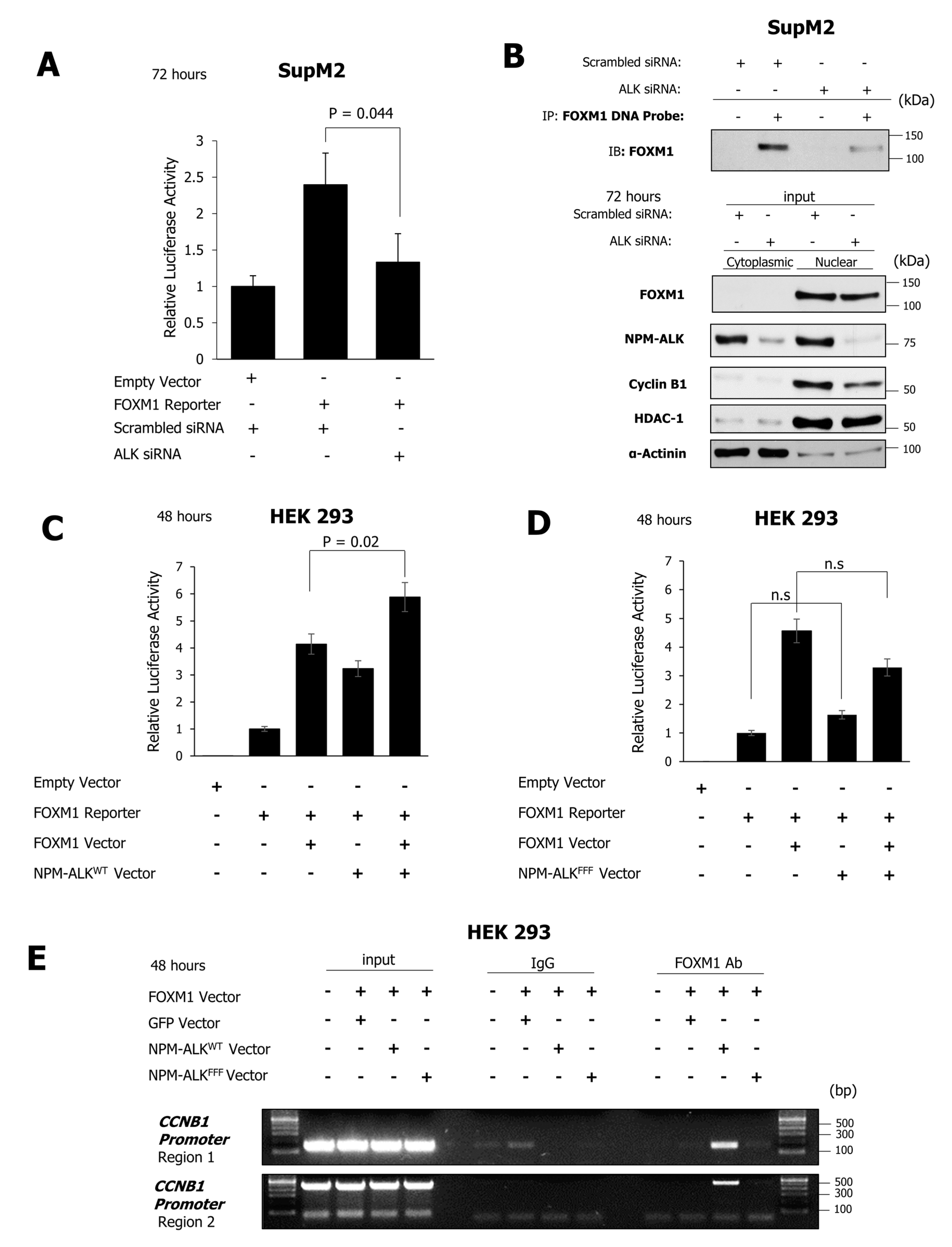
2.5. NPM-ALK Induces Transcriptional Activity of FOXM1
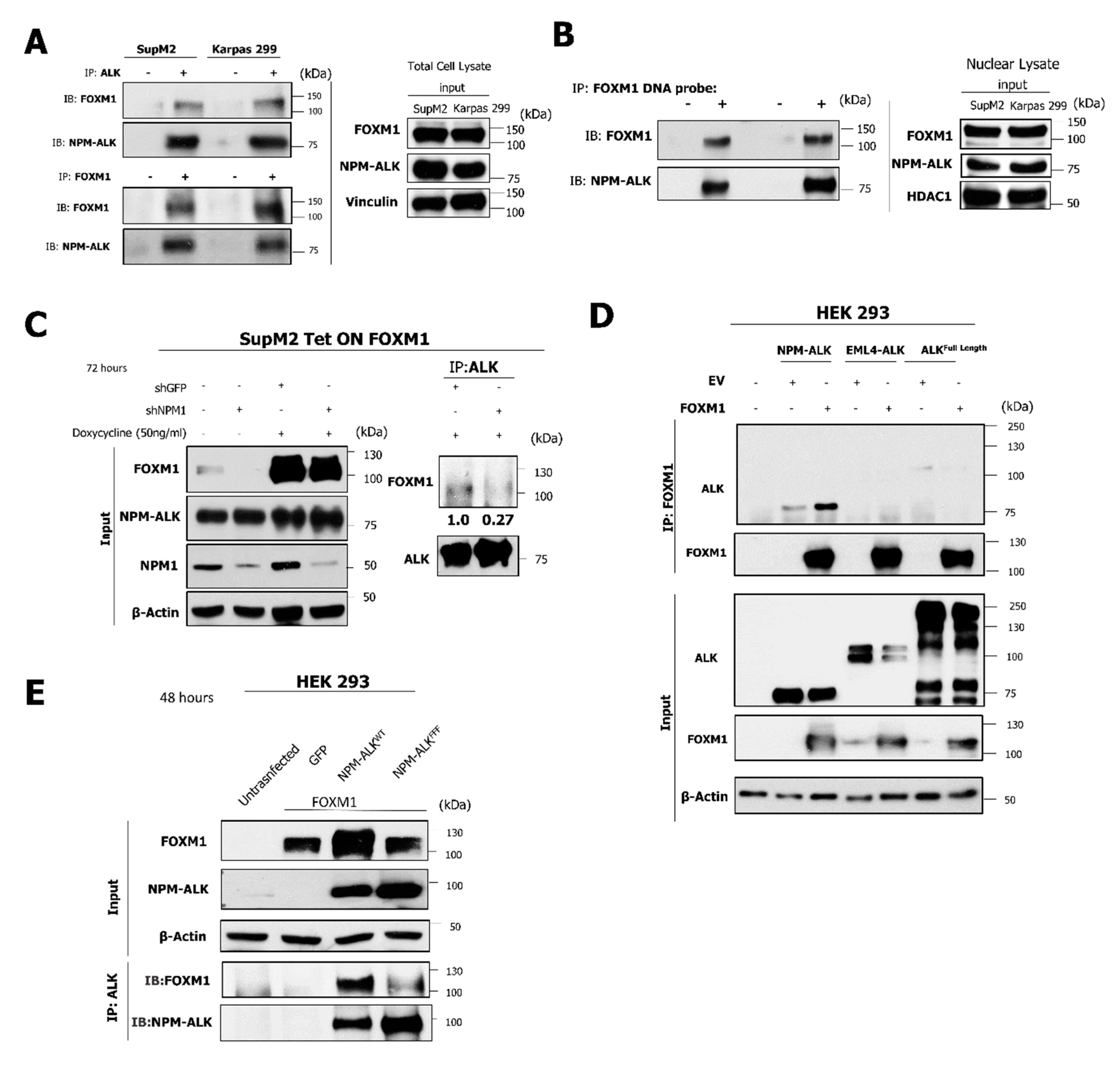
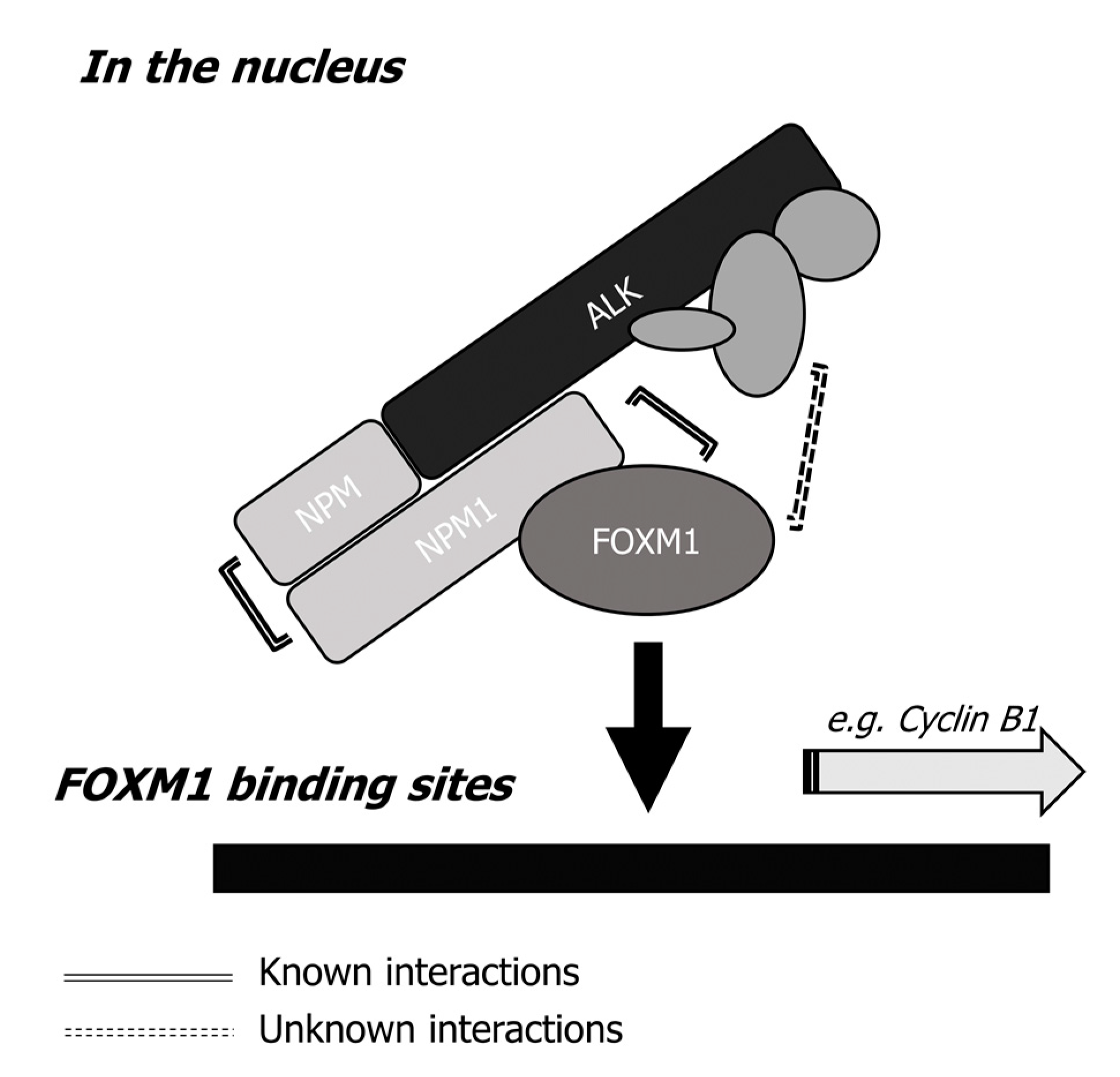
2.6. The NPM-ALK—FOXM1 Binding Is Mediated via NPM1
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Materials
4.2. Immunohistochemistry
4.3. Cell Viability
4.4. Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) and Quantitative Real Time PCR (qPCR)
4.5. Plasmids and siRNA
4.6. Transient Transfections
4.7. Lentiviral Based Transduction
4.8. Flow Cytometric Analyses
4.9. Soft Agar Colony Formation Assay
4.10. Co-Immunoprecipitation and Western Blotting
4.11. FOXM1 DNA Probe Pulldown
4.12. Luciferase Assay
4.13. Chromatin Immunoprecipitation
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koo, C.-Y.; Muir, K.W.; Lam, E.W.-F. FOXM1: From cancer initiation to progression and treatment. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2012, 1819, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Korver, W.; Roose, J.; Clevers, H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 1997, 25, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Muller, G.A.; Quaas, M.; Fischer, M.; Han, N.; Stutchbury, B.; Sharrocks, A.D.; Engeland, K. The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol. Cell. Biol. 2013, 33, 227–236. [Google Scholar] [CrossRef]
- Myatt, S.S.; Lam, E.W.-F. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Buchner, M.; Park, E.; Geng, H.M.; Klemm, L.; Flach, J.; Passegue, E.; Schjerven, H.; Melnick, A.; Paietta, E.; Kopanja, D.; et al. Identification of FOXM1 as a therapeutic target in B-cell lineage acute lymphoblastic leukaemia. Nat. Commun. 2015, 6, 6471. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, O.; Yandim, M.K.; Oren, H.; Irken, G.; Baran, Y. Targeting FOXM1 Transcription Factor In T-Cell Acute Lymphoblastic Leukemia. Blood 2013, 122, 4974. [Google Scholar]
- Wang, J.Y.; Jia, X.H.; Xing, H.Y.; Li, Y.J.; Fan, W.W.; Li, N.; Xie, S.Y. Inhibition of Forkhead box protein M1 by thiostrepton increases chemosensitivity to doxorubicin in T-cell acute lymphoblastic leukemia. Mol. Med. Rep. 2015, 12, 1457–1464. [Google Scholar] [CrossRef]
- Gu, C.; Yang, Y.; Sompallae, R.; Xu, H.; Tompkins, V.S.; Holman, C.; Hose, D.; Goldschmidt, H.; Tricot, G.; Zhan, F.; et al. FOXM1 is a therapeutic target for high-risk multiple myeloma. Leukemia 2016, 30, 873–882. [Google Scholar] [CrossRef]
- Uddin, S.; Hussain, A.R.; Ahmed, M.; Siddiqui, K.; Al-Dayel, F.; Bavi, P.; Al-Kuraya, K.S. Overexpression of FoxM1 offers a promising therapeutic target in diffuse large B-cell lymphoma. Haematologica 2012, 97, 1092–1100. [Google Scholar] [CrossRef]
- Khan, I.; Patel, A.A.; Halasi, M.; Schultz, R.; Chen, Y.H.; Aardsma, N.; Kalakota, N.; Liu, L.C.; Mahmud, N.; Gann, P.; et al. Validation of FOXM1 As a Therapeutic Target in Acute Myeloid Leukemia. Blood 2017, 130, 300. [Google Scholar]
- Amin, H.M.; Lai, R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood 2007, 110, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar] [PubMed]
- Marzec, M.; Kasprzycka, M.; Liu, X.; Raghunath, P.N.; Wlodarski, P.; Wasik, M.A. Oncogenic tyrosine kinase NPM/ALK induces activation of the MEK/ERK signaling pathway independently of c-Raf. Oncogene 2006, 26, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef]
- Gelebart, P.; Hegazy, S.A.; Wang, P.; Bone, K.M.; Anand, M.; Sharon, D.; Hitt, M.; Pearson, J.D.; Ingham, R.J.; Ma, Y.; et al. Aberrant expression and biological significance of Sox2, an embryonic stem cell transcriptional factor, in ALK-positive anaplastic large cell lymphoma. Blood Cancer J. 2012, 2, e82. [Google Scholar] [CrossRef]
- Wierstra, I. The Transcription Factor FOXM1 (Forkhead box M1): Proliferation-Specific Expression, Transcription Factor Function, Target Genes, Mouse Models, and Normal Biological Roles. Adv. Cancer Res. 2013, 118, 97–398. [Google Scholar]
- Tufekci, O.; Yandim, M.K.; Oren, H.; Irken, G.; Baran, Y. Targeting FoxM1 transcription factor in T-cell acute lymphoblastic leukemia cell line. Leuk. Res. 2015, 39, 342–347. [Google Scholar] [CrossRef][Green Version]
- Ma, R.Y.M.; Tong, T.H.K.; Cheung, A.M.S.; Tsang, A.C.C.; Leung, W.Y.; Yao, K.-M. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J. Cell Sci. 2005, 118, 795–806. [Google Scholar] [CrossRef]
- Malcolm, T.I.M.; Villarese, P.; Fairbairn, C.J.; Lamant, L.; Trinquand, A.; Hook, C.E.; Burke, G.A.A.; Brugières, L.; Hughes, K.; Payet, D.; et al. Anaplastic large cell lymphoma arises in thymocytes and requires transient TCR expression for thymic egress. Nat. Commun. 2016, 7, 10087. [Google Scholar] [CrossRef]
- Hegde, N.S.; Sanders, D.A.; Rodriguez, R.; Balasubramanian, S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat. Chem. 2011, 3, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.W.C.; Lin, S.S.W.; Tsang, A.C.C.; Tong, C.S.W.; Ching, J.C.Y.; Leung, W.Y.; Gimlich, R.; Wong, G.G.; Yao, K.M. Over-expression of FoxM1 stimulates cyclin B1 expression. FEBS Lett. 2001, 507, 59–66. [Google Scholar] [CrossRef]
- Wang, P.; Wu, F.; Zhang, J.; McMullen, T.; Young, L.C.; Ingham, R.J.; Li, L.; Lai, R. Serine phosphorylation of NPM–ALK, which is dependent on the auto-activation of the kinase activation loop, contributes to its oncogenic potential. Carcinogenesis 2010, 32, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From structure and function to disease development. BMC Mol. Biol. 2016, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Bhat, U.G.; Jagadeeswaran, R.; Halasi, M.; Gartel, A.L. Nucleophosmin interacts with FOXM1 and modulates the level and localization of FOXM1 in human cancer cells. J. Biol. Chem. 2011, 286, 41425–41433. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, P.; Gong, A.H.; Chiu, W.T.; Lee, H.T.; Colman, H.; Huang, H.; Xue, J.F.; Liu, M.G.; Wang, Y.; et al. FoxM1 promotes beta-catenin nuclear localization and controls wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef]
- Xie, Z.Q.; Tan, G.X.; Ding, M.A.; Dong, D.F.; Chen, T.H.; Meng, X.X.; Huang, X.Q.; Tan, Y.J. Foxm1 transcription factor is required for maintenance of pluripotency of P19 embryonal carcinoma cells. Nucleic Acids Res. 2010, 38, 8027–8038. [Google Scholar] [CrossRef]
- Pandit, B.; Halasi, M.; Gartel, A.L. P53 negatively regulates expression of FoxM1. Cell Cycle 2009, 8, 3425–3427. [Google Scholar] [CrossRef]
- Ahmad, A.; Wang, Z.; Kong, D.; Ali, S.; Li, Y.; Banerjee, S.; Ali, R.; Sarkar, F.H. FoxM1 down-regulation leads to inhibition of proliferation, migration and invasion of breast cancer cells through the modulation of extra-cellular matrix degrading factors. Breast Cancer Res. Treat. 2010, 122, 337–346. [Google Scholar] [CrossRef]
- Wang, Z.B.; Zheng, Y.; Park, H.J.; Li, J.; Carr, J.R.; Chen, Y.J.; Kiefer, M.M.; Kopanja, D.; Bagchi, S.; Tyner, A.L.; et al. Targeting FoxM1 effectively retards p53-null lymphoma and sarcoma. Mol. Cancer Ther. 2013, 12, 759–767. [Google Scholar] [CrossRef]
- Siraj, A.K.; Hussain, A.R.; Ahmed, M.; Ahmed, S.O.; Bu, R.; Al-Dayel, F.; Bavi, P.; Uddin, S.; Al-Kuraya, K.S. FoxM1 expression and its association with matrix metalloproteinases in diffuse large B-cell lymphoma. Cancer Res. 2011, 71, 1492. [Google Scholar]
- Khan, I.; Halasi, M.; Patel, A.; Schultz, R.; Kalakota, N.; Chen, Y.H.; Aardsma, N.; Liu, L.; Crispino, J.D.; Mahmud, N.; et al. FOXM1 contributes to treatment failure in acute myeloid leukemia. JCI Insight 2018, 3, 121583. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Bigerna, B.; Fizzotti, M.; Pulford, K.; Pileri, S.A.; Delsol, G.; Carbone, A.; Paulli, M.; Magrini, U.; Menestrina, F.; et al. ALK expression defines a distinct group of T/null lymphomas (“ALK lymphomas”) with a wide morphological spectrum. Am. J. Pathol. 1998, 153, 875–886. [Google Scholar] [CrossRef]
- Falini, B.; Martelli, M.P. Anaplastic large cell lymphoma: Changes in the World Health Organization classification and perspectives for targeted therapy. Haematologica 2009, 94, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Chiang, L.; He, B.; Zhao, Y.Y.; Winoto, A. FoxM1, a Forkhead Transcription Factor Is a Master Cell Cycle Regulator for Mouse Mature T Cells but Not Double Positive Thymocytes. PLoS ONE 2010, 5, e9229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.C.; Gong, A.H.; Wei, P.; Zhou, A.D.; Yao, J.; Yuan, Y.; Lang, F.; Rao, G.; Huang, S.Y. FoxM1 drives a feed-forward STAT3-activation signaling loop to promote the self-renewal and tumorigenicity of glioblastoma stem cells. Cancer Res. 2015, 75, 2337–2348. [Google Scholar]
- Wang, I.C.; Chen, Y.J.; Hughes, D.E.; Ackerson, T.; Major, M.L.; Kalinichenko, V.V.; Costa, R.H.; Raychaudhuri, P.; Tyner, A.L.; Lau, L.F. FoxM1 regulates transcription of JNK1 to promote the G(1)/S transition and tumor cell invasiveness. J. Biol. Chem. 2008, 283, 20770–20778. [Google Scholar] [CrossRef]
- Khan, I.; Zia, M.; Halasi, M.; Gann, P.; Gaitonde, S.; Gartel, A. FOXM1 Binds Nucleophosmin in AML and Confers Resistance to Chemotherapy. Blood 2015, 126, 2467. [Google Scholar]
- Ceccon, M.; Merlo, M.E.B.; Mologni, L.; Poggio, T.; Varesio, L.M.; Menotti, M.; Bombelli, S.; Rigolio, R.; Manazza, A.D.; Di Giacomo, F.; et al. Excess of NPM-ALK oncogenic signaling promotes cellular apoptosis and drug dependency. Oncogene 2016, 35, 3854–3865. [Google Scholar] [CrossRef]
- Sabir, S.R.; Yeoh, S.; Jackson, G.; Bayliss, R. EML4-ALK Variants: Biological and Molecular Properties, and the Implications for Patients. Cancers 2017, 9, 118. [Google Scholar] [CrossRef]
- Wellstein, A. ALK receptor activation, ligands and therapeutic targeting in glioblastoma and in other cancers. Front. Oncol. 2012, 2, 192. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.M.; Chan, S.M.; Minden, M.D.; Murphy, T.; Shlush, L.I.; Schimmer, A.D. Biological and clinical consequences of NPM1 mutations in AML. Leukemia 2017, 31, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Boone, D.; Hann, S.R. Nucleophosmin interacts directly with c-Myc and controls c-Myc-induced hyperproliferation and transformation. Proc. Natl. Acad. Sci. USA 2008, 105, 18794–18799. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kato, M.; Nagata, K.; Okuwaki, M. Efficient DNA binding of NF-κB requires the chaperone-like function of NPM1. Nucleic Acids Res. 2017, 45, 3707–3723. [Google Scholar] [CrossRef]
- Gelebart, P.; Anand, M.; Armanious, H.; Peters, A.C.; Dien Bard, J.; Amin, H.M.; Lai, R. Constitutive activation of the Wnt canonical pathway in mantle cell lymphoma. Blood 2008, 112, 5171–5179. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Molavi, O.; Zhang, H.; Gupta, N.; Alshareef, A.; Bone, K.M.; Gopal, K.; Wu, F.; Lewis, J.T.; Douglas, D.N.; et al. STAT1 is phosphorylated and downregulated by the oncogenic tyrosine kinase NPM-ALK in ALK-positive anaplastic large-cell lymphoma. Blood 2015, 126, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Barger, C.J.; Zhang, W.; Hillman, J.; Stablewski, A.B.; Higgins, M.J.; Vanderhyden, B.C.; Odunsi, K.; Karpf, A.R. Genetic determinants of FOXM1 overexpression in epithelial ovarian cancer and functional contribution to cell cycle progression. Oncotarget 2015, 6, 27613–27627. [Google Scholar] [CrossRef]
- Barger, C.J.; Branick, C.; Chee, L.; Karpf, A.R. Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers 2019, 11, 251. [Google Scholar] [CrossRef]
- Lee, J.; Sadelain, M.; Brentjens, R. Retroviral transduction of murine primary T lymphocytes. Methods Mol. Biol. (Clifton N.J.) 2009, 506, 83–96. [Google Scholar]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The Soft Agar Colony Formation Assay. JoVE 2014, e51998. [Google Scholar] [CrossRef]
- Wu, K.K. Analysis of Protein-DNA Binding by Streptavidin-Agarose Pulldown. In Gene Mapping, Discovery, and Expression: Methods and Protocols; Bina, M., Ed.; Humana Press: Totowa, NJ, USA, 2006; pp. 281–290. [Google Scholar]
- Ketola, K.; Bishop, J.; Nip, K.M.; Kim, S.; Ladan, F.; Gleave, M.; Zoubeidi, A. Inhibition of FOXM1 targets both high and low PSA expressing prostate cancer cells resistant to Enzalutamide. Cancer Res. 2014, 74. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, M.; Li, J.; Huang, Y.-H.; Almowaled, M.; Barger, C.J.; Karpf, A.R.; Wang, P.; Chen, W.; Turner, S.D.; Lai, R. NPM-ALK Is a Key Regulator of the Oncoprotein FOXM1 in ALK-Positive Anaplastic Large Cell Lymphoma. Cancers 2019, 11, 1119. https://doi.org/10.3390/cancers11081119
Haque M, Li J, Huang Y-H, Almowaled M, Barger CJ, Karpf AR, Wang P, Chen W, Turner SD, Lai R. NPM-ALK Is a Key Regulator of the Oncoprotein FOXM1 in ALK-Positive Anaplastic Large Cell Lymphoma. Cancers. 2019; 11(8):1119. https://doi.org/10.3390/cancers11081119
Chicago/Turabian StyleHaque, Moinul, Jing Li, Yung-Hsing Huang, Meaad Almowaled, Carter J. Barger, Adam R. Karpf, Peng Wang, Will Chen, Suzanne D. Turner, and Raymond Lai. 2019. "NPM-ALK Is a Key Regulator of the Oncoprotein FOXM1 in ALK-Positive Anaplastic Large Cell Lymphoma" Cancers 11, no. 8: 1119. https://doi.org/10.3390/cancers11081119
APA StyleHaque, M., Li, J., Huang, Y.-H., Almowaled, M., Barger, C. J., Karpf, A. R., Wang, P., Chen, W., Turner, S. D., & Lai, R. (2019). NPM-ALK Is a Key Regulator of the Oncoprotein FOXM1 in ALK-Positive Anaplastic Large Cell Lymphoma. Cancers, 11(8), 1119. https://doi.org/10.3390/cancers11081119