High FREM2 Gene and Protein Expression Are Associated with Favorable Prognosis of IDH-WT Glioblastomas

 ,
,  ,
, 

Abstract
1. Introduction
2. Results
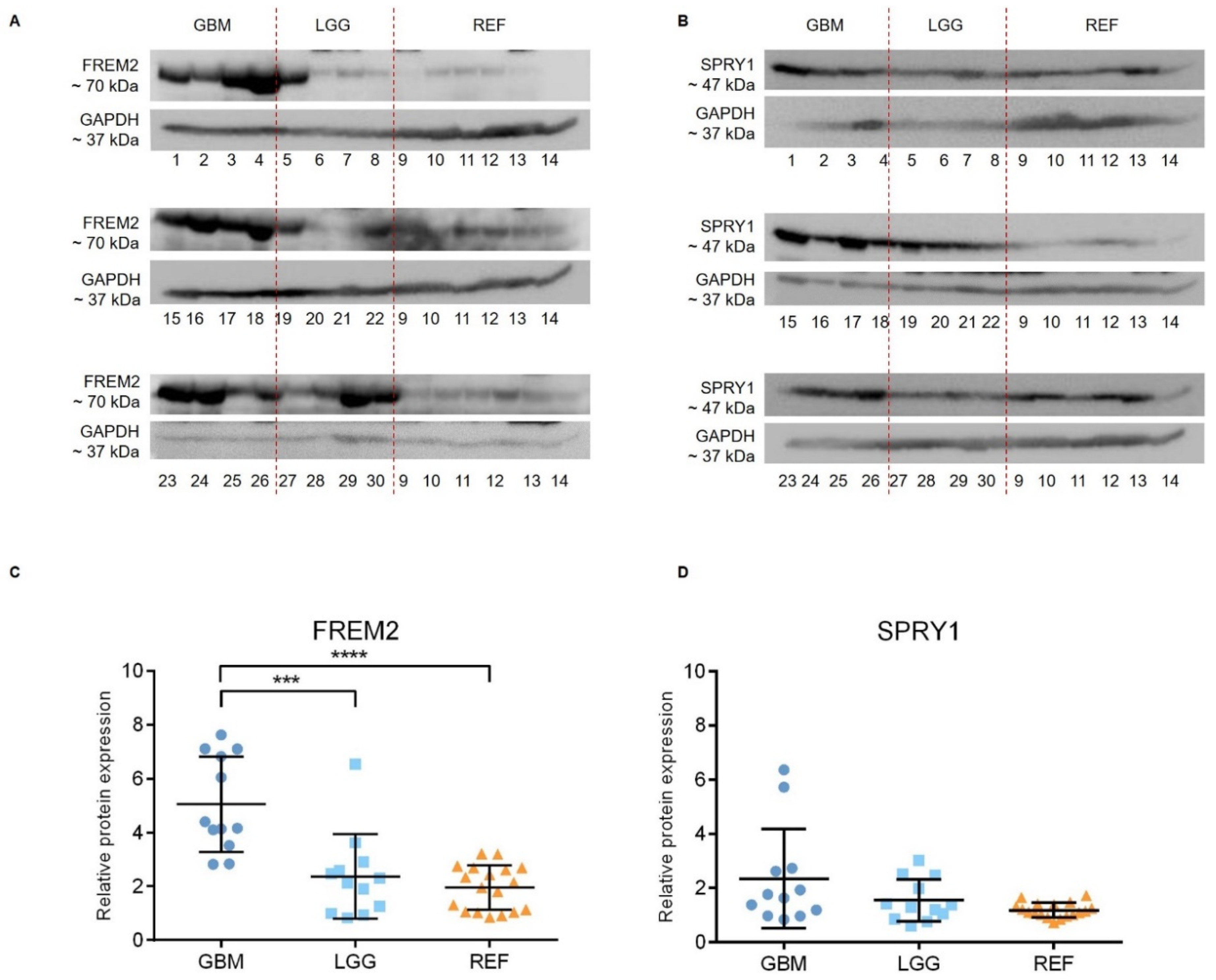
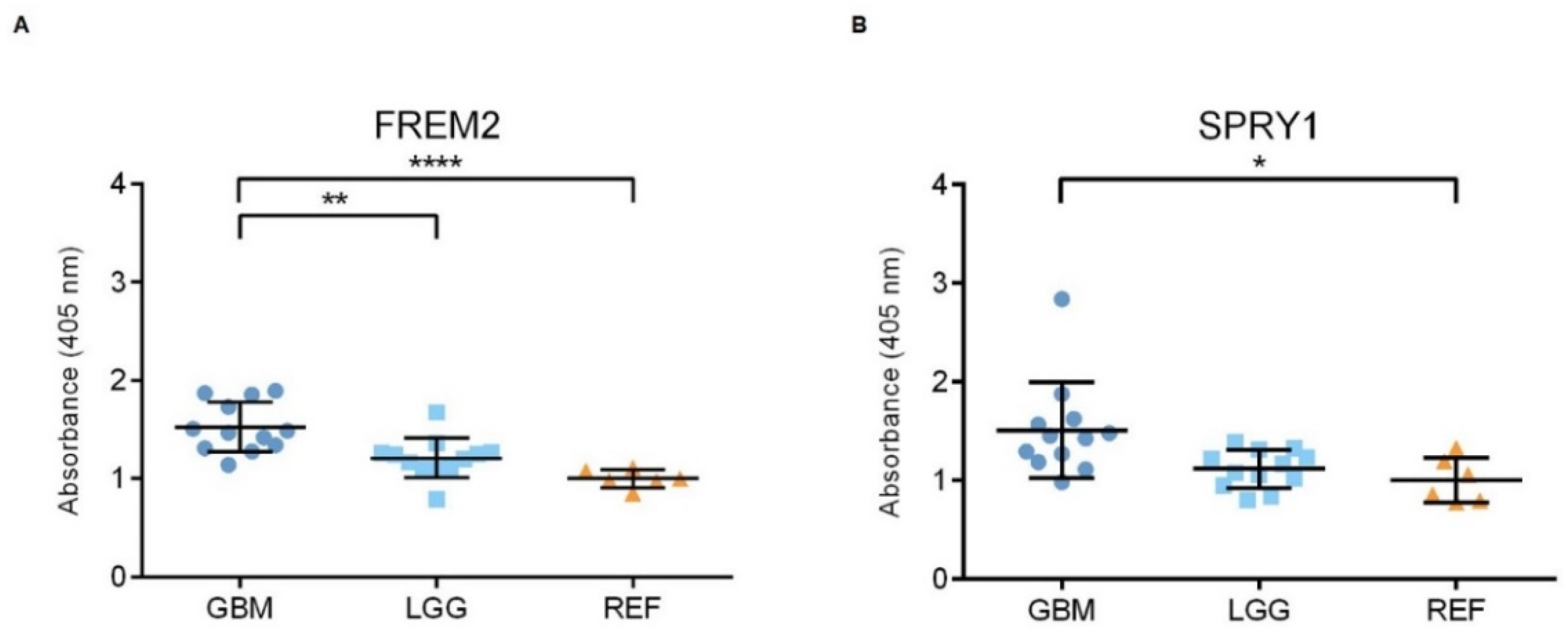
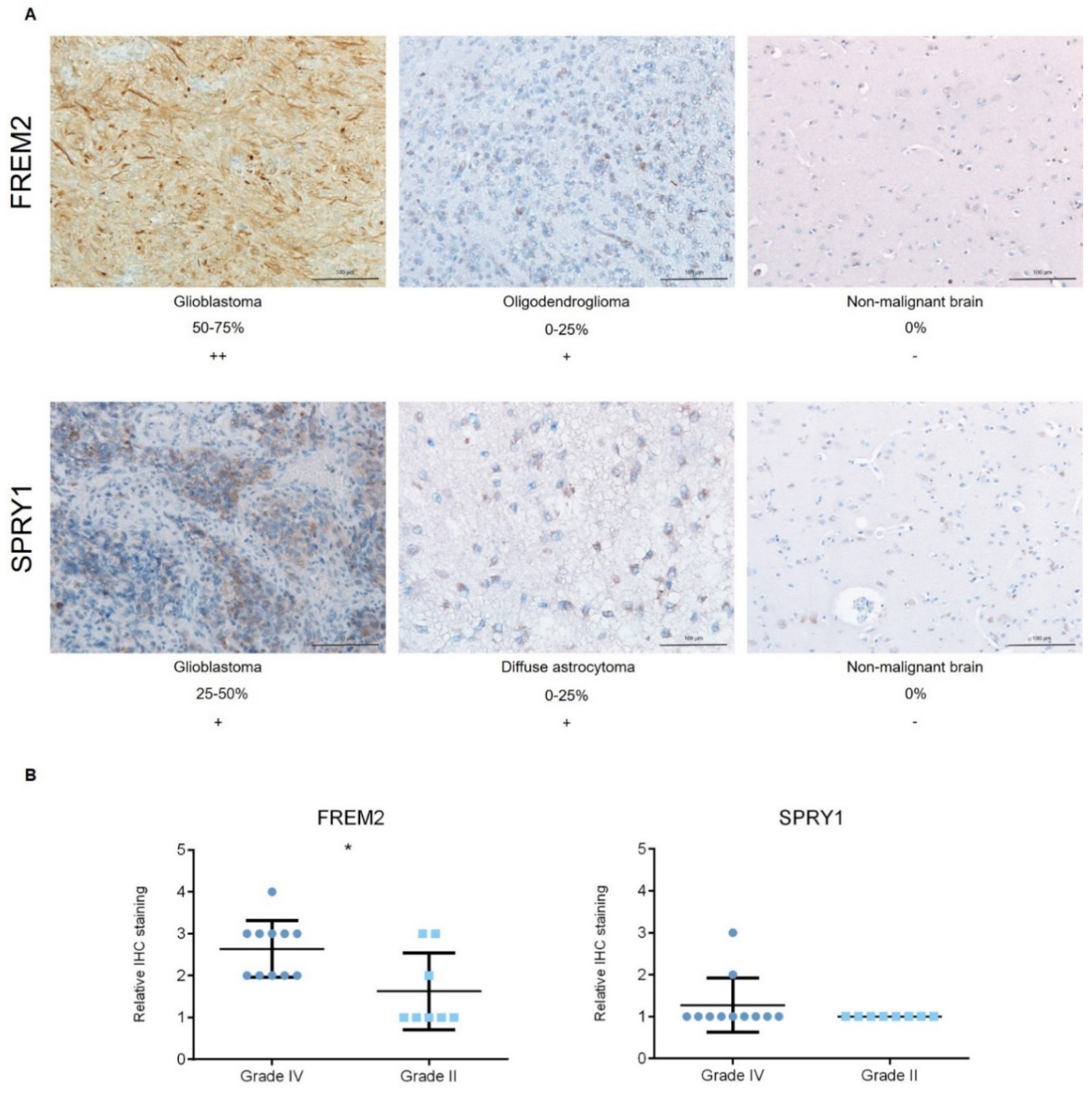
2.1. The FREM2 Protein Shows Higher Levels in Glioblastomas Versus Lower Grade Gliomas
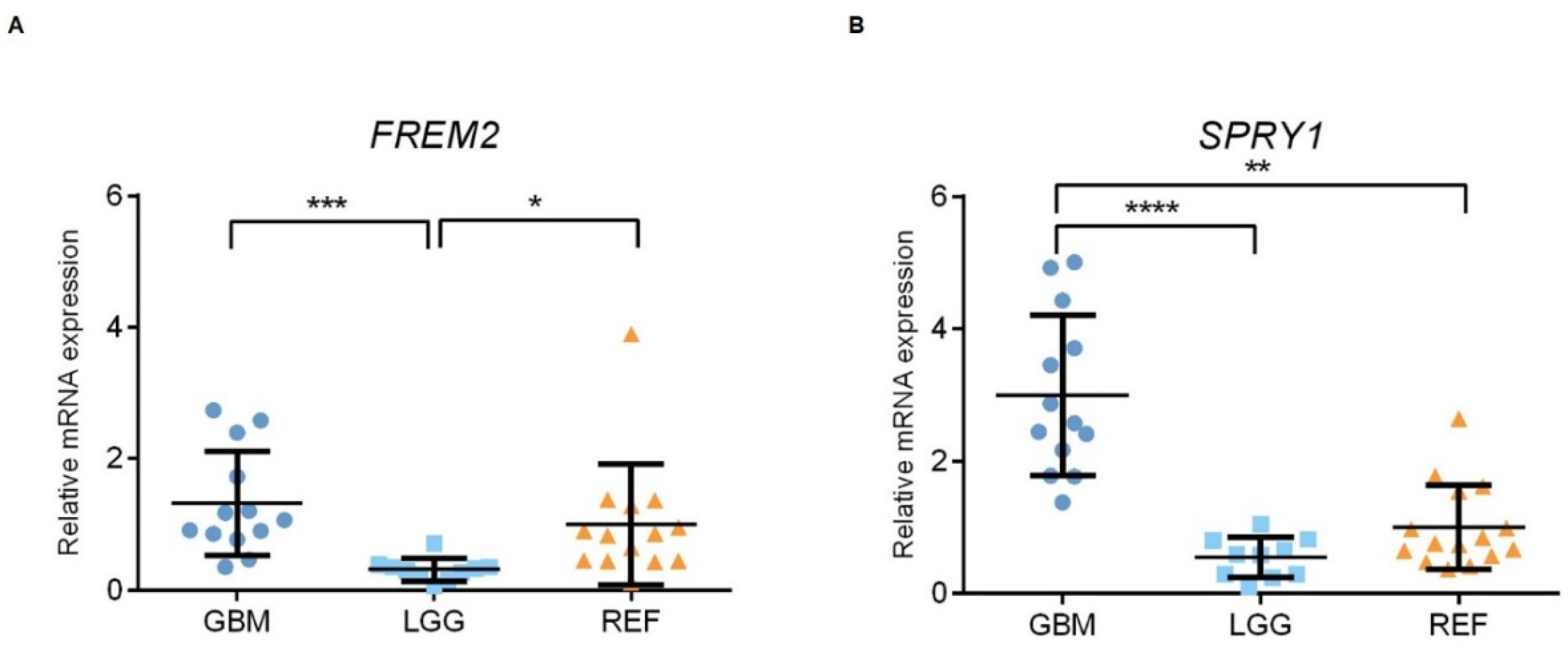
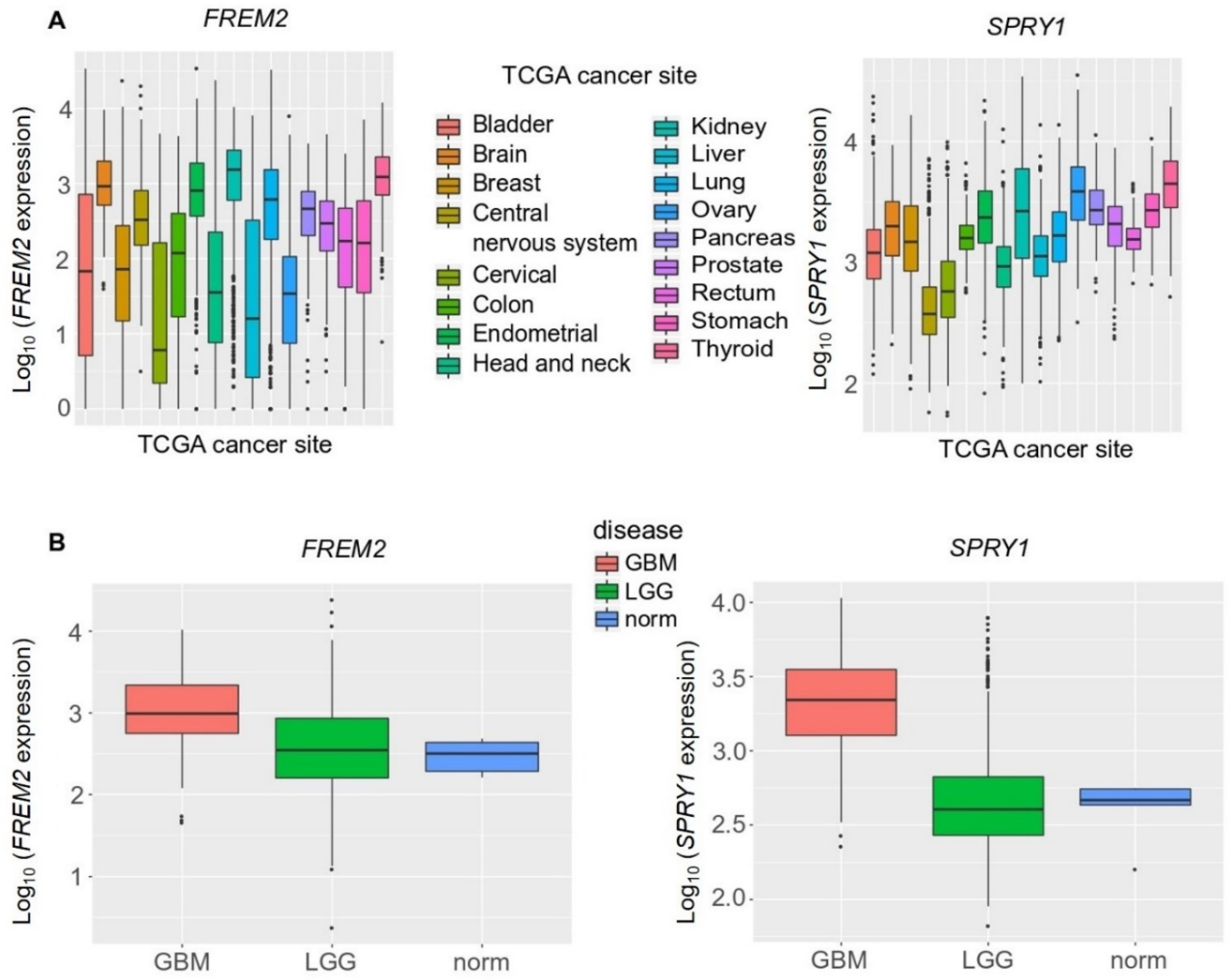
2.2. The FREM2 and SPRY1 Genes Show Higher Expression in Glioblastomas Versus Lower Grade Gliomas
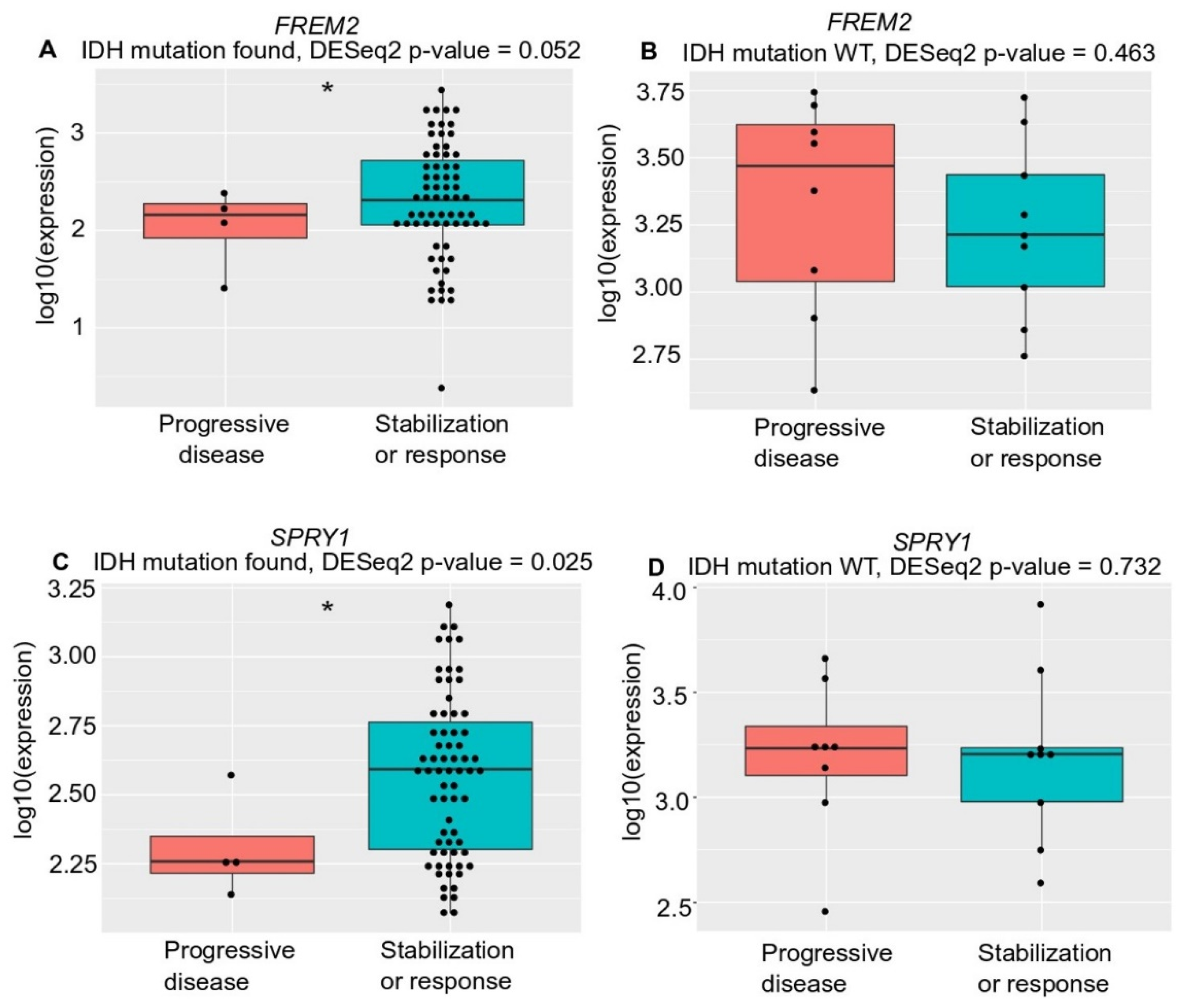
2.3. FREM2 and SPRY1 Show Lower Expression in IDH-Mutant Low-Grade Gliomas that Progress After First-Line Temozolomide Treatment
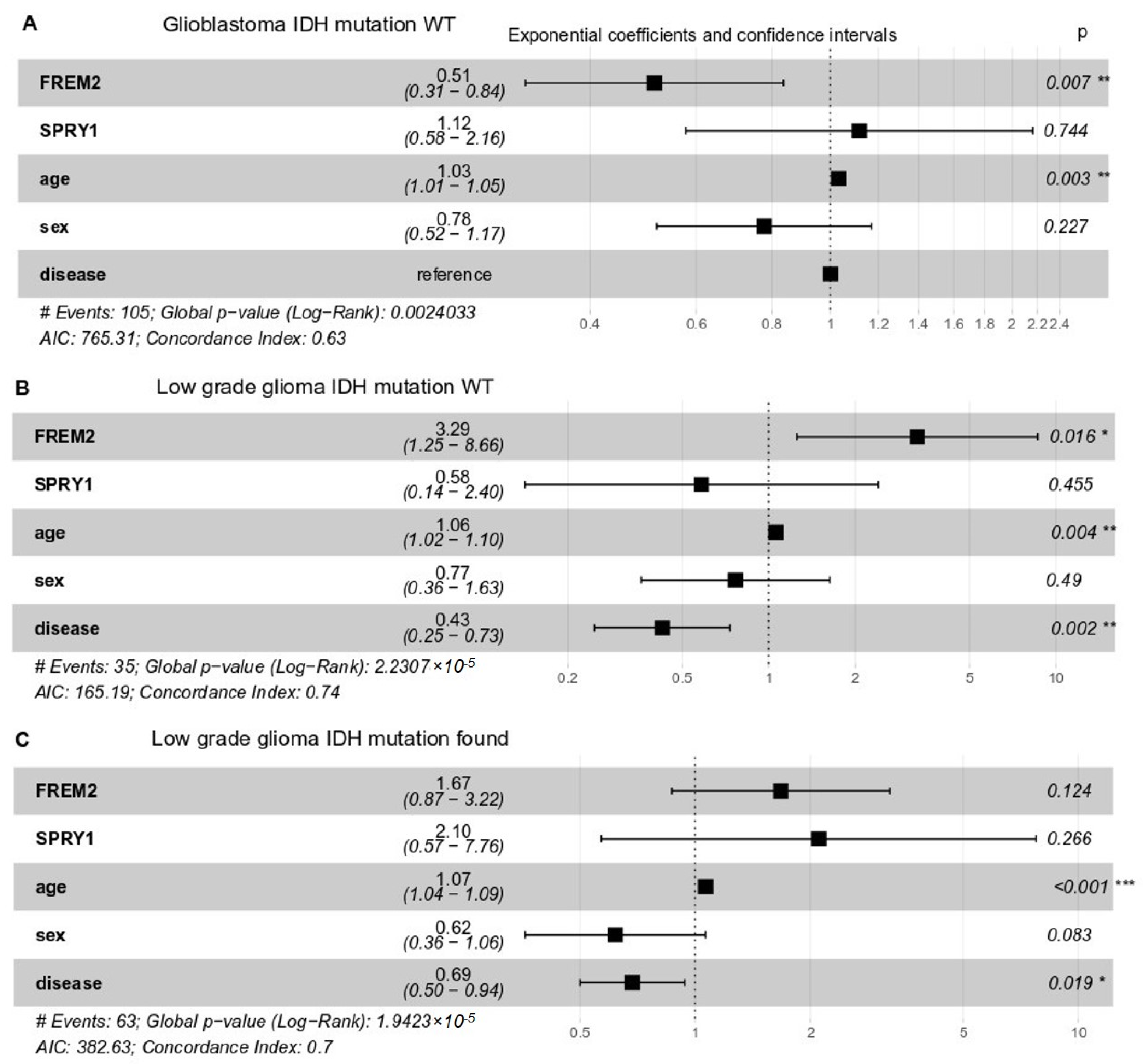
2.4. High FREM2 Expression Is Positively Associated with IDH-WT Glioblastoma Patient Survival and Negatively with IDH-WT Low Grade Glioma Patient Survival
3. Discussion
3.1. FRAS1-Related Extracellular Matrix Protein 2—FREM2
3.2. Sprouty RTK Signaling Antagonist 1—SPRY1
4. Materials and Methods
4.1. Ethics Statement
4.2. Tissue Samples
4.3. Proteomic Analysis
4.3.1. Protein Isolation
4.3.2. Immunoblotting
4.3.3. Enzyme-Linked Immunosorbent Assay
4.3.4. Immunohistochemistry
4.4. Transcriptomic Analysis
4.4.1. RNA Extraction
4.4.2. Quantitative Real-Time Polymerase Chain Reaction
4.5. Statistical Analysis
4.6. Bioinformatic Analysis
In-Silico Analysis Using the Cancer Genome Atlas Datasets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fawzy, M.S.; Badran, D.I.; Al Ageeli, E.; Al-Qahtani, S.A.M.; Alghamdi, S.A.; Helal, G.M.; Toraih, E.A. Longevity-Related Gene Transcriptomic Signature in Glioblastoma Multiforme. Oxid. Med. Cell. Longev. 2018, 2018, 8753063. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neuro-Oncol. 2012, 107, 359–364. [Google Scholar] [CrossRef]
- Noroxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, e000144. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.; Kanu, O.O.; Mehta, A.I.; Di, C.; Lin, N.; Mattox, A.K.; Bigner, D.D. Glioblastoma multiforme: A review of where we have been and where we are going. Expert Opin. Investig. Drugs 2009, 18, 1061–1083. [Google Scholar] [CrossRef] [PubMed]
- Gately, L.; McLachlan, S.A.; Philip, J.; Ruben, J.; Dowling, A. Long-term survivors of glioblastoma: A closer look. J. Neurooncol. 2018, 136, 155–162. [Google Scholar] [CrossRef]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.K.; Reijneveld, J.C.; Enting, R.H.; Bienfait, H.P.; Robe, P.; Baumert, B.G.; Visser, O. Dutch Society for Neuro-Oncology. Changing incidence and improved survival of gliomas. Eur. J. Cancer 2014, 50, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R. Cell- and peptide-based immunotherapeutic approaches for glioma. Trends Mol. Med. 2008, 14, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Inda, M.M.; Cavenee, W.K.; Furnari, F.B. Heterogeneity maintenance in glioblastoma: A social network. Cancer Res. 2011, 71, 4055–4060. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Gruber, M.L.; Buster, W.P. Temozolomide in combination with irinotecan for treatment of recurrent malignant glioma. Am. J. Clin. Oncol. 2004, 27, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Veliz, I.; Loo, Y.; Castillo, O.; Karachaliou, N.; Nigro, O.; Rosell, R. Advances and challenges in the molecular biology and treatment of glioblastoma-is there any hope for the future? Ann. Transl. Med. 2015, 3, 7. [Google Scholar]
- Aum, D.J.; Kim, D.H.; Beaumont, T.L.; Leuthardt, E.C.; Dunn, G.P.; Kim, A.H. Molecular and cellular heterogeneity: The hallmark of glioblastoma. Neurosurg. Focus 2014, 37, E11. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.; Iwama, T.; Park, D.M. The evidence of glioblastoma heterogeneity. Sci. Rep. 2015, 5, 7979. [Google Scholar] [CrossRef]
- Eder, K.; Kalman, B. Molecular heterogeneity of glioblastoma and its clinical relevance. Pathol. Oncol. Res. 2014, 20, 777–787. [Google Scholar] [CrossRef]
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856. [Google Scholar] [CrossRef]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, T.; Jiang, Y.; Xu, H.; Yan, Y.; Fu, D.; Chen, J. The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia 2015, 17, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, H.; Yan, W.; Yang, P.; Bao, Z.; Zhang, C.; Jiang, T.; You, Y. Genetic and clinical characteristics of primary and secondary glioblastoma is associated with differential molecular subtype distribution. Oncotarget 2015, 6, 7318–7324. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Oh, J.E.; Ohta, T.; Nonoguchi, N.; Satomi, K.; Capper, D.; Pierscianek, D.; Sure, U.; Vital, A.; Paulus, W.; Mittelbronn, M.; et al. Genetic Alterations in Gliosarcoma and Giant Cell Glioblastoma. Brain Pathol. 2016, 26, 517–522. [Google Scholar] [CrossRef]
- Khanna, G.; Pathak, P.; Suri, V.; Sharma, M.C.; Chaturvedi, S.; Ahuja, A.; Bhardwaj, M.; Garg, A.; Sarkar, C.; Sharma, R. Immunohistochemical and molecular genetic study on epithelioid glioblastoma: Series of seven cases with review of literature. Pathol. Res. Pract. 2018, 214, 679–685. [Google Scholar] [CrossRef]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Czapski, B.; Baluszek, S.; Herold-Mende, C.; Kaminska, B. Clinical and immunological correlates of long term survival in glioblastoma. Contemp Oncol. 2018, 22, 81–85. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A., 3rd; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharm. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.J.; Ali, S.; Qadir, M.G.; De La Fuente, M.I.; Ivan, M.E.; Komotar, R.J. The role of bevacizumab in the treatment of glioblastoma. J. Neuro-Oncol. 2017, 133, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Vidak, M.; Jovcevska, I.; Samec, N.; Zottel, A.; Liovic, M.; Rozman, D.; Dzeroski, S.; Juvan, P.; Komel, R. Meta-Analysis and Experimental Validation Identified FREM2 and SPRY1 as New Glioblastoma Marker Candidates. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Biswas, N.K.; Chandra, V.; Sarkar-Roy, N.; Das, T.; Bhattacharya, R.N.; Tripathy, L.N.; Basu, S.K.; Kumar, S.; Das, S.; Chatterjee, A.; et al. Variant allele frequency enrichment analysis in vitro reveals sonic hedgehog pathway to impede sustained temozolomide response in GBM. Sci. Rep. 2015, 5, 7915. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, E.; Chiotaki, R.; Chalepakis, G. The role of Fras1/Frem proteins in the structure and function of basement membrane. Int. J. Biochem. Cell Biol. 2011, 43, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Nagaishi, M.; Kim, Y.H.; Mittelbronn, M.; Giangaspero, F.; Paulus, W.; Brokinkel, B.; Vital, A.; Tanaka, Y.; Nakazato, Y.; Legras-Lachuer, C.; et al. Amplification of the STOML3, FREM2, and LHFP genes is associated with mesenchymal differentiation in gliosarcoma. Am. J. Pathol. 2012, 180, 1816–1823. [Google Scholar] [CrossRef]
- Arita, H.; Yamasaki, K.; Matsushita, Y.; Nakamura, T.; Shimokawa, A.; Takami, H.; Tanaka, S.; Mukasa, A.; Shirahata, M.; Shimizu, S.; et al. A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroups of newly diagnosed glioblastomas. Acta Neuropathol. Commun. 2016, 4, 79. [Google Scholar] [CrossRef]
- Choi, S.; Yu, Y.; Grimmer, M.R.; Wahl, M.; Chang, S.M.; Costello, J.F. Temozolomide-associated hypermutation in gliomas. Neuro-Oncol. 2018, 20, 1300–1309. [Google Scholar] [CrossRef]
- Muscat, A.M.; Wong, N.C.; Drummond, K.J.; Algar, E.M.; Khasraw, M.; Verhaak, R.; Field, K.; Rosenthal, M.A.; Ashley, D.M. The evolutionary pattern of mutations in glioblastoma reveals therapy-mediated selection. Oncotarget 2018, 9, 7844–7858. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Zhao, Q.; Yan, J.; Kong, L.Y.; Gabrusiewicz, K.; Hong, S.; Xia, X.; Heimberger, A.B.; Li, S. Cell surface vimentin-targeted monoclonal antibody 86C increases sensitivity to temozolomide in glioma stem cells. Cancer Lett. 2018, 433, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lan, Y.; Zhang, D.; Wang, K.; Wang, Y.; Hua, Z.C. SPRY1 promotes the degradation of uPAR and inhibits uPAR-mediated cell adhesion and proliferation. Am. J. Cancer Res. 2014, 4, 683–697. [Google Scholar] [PubMed]
- Mekkawy, A.H.; Pourgholami, M.H.; Morris, D.L. Human Sprouty1 suppresses growth, migration, and invasion in human breast cancer cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 5037–5048. [Google Scholar]
- Yuan, Z.S.; Cao, Y.; Li, Z.Y. IGFBP2 induces SPRY1 expression via NF-kappaB signaling pathway in glioblastoma multiforme (GBM). Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5072–5080. [Google Scholar] [PubMed]
- Lo, T.L.; Fong, C.W.; Yusoff, P.; McKie, A.B.; Chua, M.S.; Leung, H.Y.; Guy, G.R. Sprouty and cancer: The first terms report. Cancer Lett. 2006, 242, 141–150. [Google Scholar] [CrossRef]
- Zhang, W.; Lv, Y.; Xue, Y.; Wu, C.; Yao, K.; Zhang, C.; Jin, Q.; Huang, R.; Li, J.; Sun, Y.; et al. Co-expression modules of NF1, PTEN and sprouty enable distinction of adult diffuse gliomas according to pathway activities of receptor tyrosine kinases. Oncotarget 2016, 7, 59098–59114. [Google Scholar] [CrossRef]
- Aldaz, B.; Sagardoy, A.; Nogueira, L.; Guruceaga, E.; Grande, L.; Huse, J.T.; Aznar, M.A.; Diez-Valle, R.; Tejada-Solis, S.; Alonso, M.M.; et al. Involvement of miRNAs in the differentiation of human glioblastoma multiforme stem-like cells. PLoS ONE 2013, 8, e77098. [Google Scholar] [CrossRef]
- Jovcevska, I.; Zupanec, N.; Urlep, Z.; Vranic, A.; Matos, B.; Stokin, C.L.; Muyldermans, S.; Myers, M.P.; Buzdin, A.A.; Petrov, I.; et al. Differentially expressed proteins in glioblastoma multiforme identified with a nanobody-based anti-proteome approach and confirmed by OncoFinder as possible tumor-class predictive biomarker candidates. Oncotarget 2017, 8, 44141–44158. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Grube, S.; Gottig, T.; Freitag, D.; Ewald, C.; Kalff, R.; Walter, J. Selection of suitable reference genes for expression analysis in human glioma using RT-qPCR. J. Neuro-Oncol. 2015, 123, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Valente, V.; Teixeira, S.A.; Neder, L.; Okamoto, O.K.; Oba-Shinjo, S.M.; Marie, S.K.; Scrideli, C.A.; Paco-Larson, M.L.; Carlotti, C.G., Jr. Selection of suitable housekeeping genes for expression analysis in glioblastoma using quantitative RT-PCR. BMC Mol. Biol. 2009, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ishikawa, T.; Michiue, T.; Zhu, B.L.; Guan, D.W.; Maeda, H. Stability of endogenous reference genes in postmortem human brains for normalization of quantitative real-time PCR data: Comprehensive evaluation using geNorm, NormFinder, and BestKeeper. Int. J. Leg. Med. 2012, 126, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Reaction Intensity | Positive Cells (%) | ||
|---|---|---|---|---|
| FREM2 | SPRY1 | FREM2 | SPRY1 | |
| Oligodendroglioma | + | + | 0–25 | 0–25 |
| Diffuse astrocytoma | + | + | 0–25 | 0–25 |
| Primary glioblastoma | ++ | + | 25–50 | 0–25 |
| Glioblastoma | ++ | ++ | 75–100 | 25–50 |
| Oligodendroglioma | + | + | 0–25 | 0–25 |
| Secondary gliosarcoma | ++ | + | 50–75 | 0–25 |
| Primary glioblastoma | ++ | + | 25–50 | 0–25 |
| Primary glioblastoma | ++ | + | 25–50 | 0–25 |
| Glioblastoma | ++ | + | 50–75 | 0–25 |
| Oligodendroglioma | + | + | 0–25 | 0–25 |
| Epitheloid glioblastoma | ++ | ++ | 50–75 | 50–75 |
| Recurrent glioblastoma | ++ | + | 25–50 | 0–25 |
| Anaplastic astrocytoma | ++ | + | 0–25 | 0–25 |
| Glioblastoma | ++ | + | 25–50 | 0–25 |
| Glioblastoma | ++ | + | 50–75 | 0–25 |
| Glioblastoma | +++ | + | 50–75 | 0–25 |
| Diffuse astrocytoma | ++ | + | 50–75 | 0–25 |
| Anaplastic astrocytoma | ++ | + | 25–50 | 0–25 |
| Diffuse astrocytoma | ++ | + | 25–50 | 0–25 |
| Oligodendroglioma | ++ | + | 0–25 | 0–25 |
| Diffuse astrocytoma | + | + | 50–75 | 0–25 |
| Feature | Detail | Glioma Grade (WHO) | ||
|---|---|---|---|---|
| II | III | IV | ||
| Number of samples | (N) | 11 | 2 | 12 |
| Gender (n) | Female (n) | 4 | 4 | |
| Male (n) | 7 | 2 | 8 | |
| Age range | (years) | 25–53 | 29–34 | 41–81 |
| Karnofsky performance scale | (%) | 60–100 | 70–90 | 40–100 |
| Overall survival | Median (months) | 21 | 15 | |
| Patients still alive | (n) | 8 | 2 | 3 |
| Diagnosis (n) | Oligodendroglioma | 2 | ||
| Diffuse astrocytoma | 9 | |||
| Anaplastic astrocytoma | 2 | |||
| Glioblastoma | 10 | |||
| Giant cell glioblastoma | 1 | |||
| Gliosarcoma | 1 | |||
| Anatomical location (n) | Parietal lobe | 1 (right) | 1 (right) | |
| Frontal lobe | 4 (right) 2 (left) | 1 (right) | 1 (right) 3 (left) | |
| Insular cortex | 2 (right) | 1 (left) | ||
| Temporal lobe | 2 (right) | 2 (right) 3 (left) | ||
| Occipital lobe | 1 (right) | |||
| Parietal occipital lobe | 1 (right) | |||
| 1p/19q codeletion (n) | Positive | 2 | ||
| Negative | 5 | 1 | ||
| 19q deletion | 1 | |||
| N/A | 3 | 2 | 11 | |
| IDH R132H status (n) | Wild-type | 2 | 12 | |
| Mutated | 8 | 2 | ||
| N/A | 1 | |||
| ATRX (n) | Loss | 6 | 2 | 1 |
| No loss | 3 | 9 | ||
| Inconclusive | 1 | |||
| N/A | 1 | 2 | ||
| TP53 (n) | Wild-type | 8 | 8 | |
| Mutated | 1 | 2 | 4 | |
| Inconclusive | 1 | |||
| N/A | 1 | |||
| Patient Number | Anatomical Location | NO of Samples |
|---|---|---|
| 1 | Hippocampus | 1 |
| Periventricular zone | 1 | |
| 2 | Subventricular zone | 1 |
| Hippocampus | 1 | |
| 3 | Brain | 3 |
| 4 | Hippocampus | 2 |
| Subventricular zone | 1 | |
| 5 | Hippocampus | 1 |
| Periventricular zone | 1 | |
| 6 | Periventricular zone | 1 |
| Hippocampus | 1 | |
| 7 | Brain | 3 |
| 8 | Subventricular zone | 1 |
| 9 | Periventricular zone | 1 |
| Hippocampus | 1 | |
| 10 | Hippocampus | 1 |
| Periventricular zone | 1 |
| Gene | Primer Pair (5′ → 3′) |
|---|---|
| RPL13A | F: CCT GGA GGA GAA GAG GAA AGA GA R: TTG AGG ACC TCT GTG TAT TTG TCA A |
| CYC1 | F: GAG GTG GAG GTT CAA GAC GG R: TAG CTC GCA CGA TGT AGC TG |
| FREM2 | F: TGA GCC AAC TGT GTT TAT TC R: GTA TAA CAG ACC ACC ATC AAC |
| SPRY1 | F: CTT TGC ATT AGG ATT TCA GAT G R: GGA TCA CAA CTA ACG AAC TG |
| Cancer Site | No of Samples |
|---|---|
| Rectum | 172 |
| Pancreas | 182 |
| Cervical | 309 |
| Ovary | 375 |
| Stomach | 407 |
| Liver | 424 |
| Bladder | 430 |
| Central nervous system | 676 |
| Head and neck | 546 |
| Prostate | 551 |
| Thyroid | 568 |
| Endometrium | 570 |
| Kidney | 1017 |
| Lung | 1135 |
| Breast | 1216 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jovčevska, I.; Zottel, A.; Šamec, N.; Mlakar, J.; Sorokin, M.; Nikitin, D.; Buzdin, A.A.; Komel, R. High FREM2 Gene and Protein Expression Are Associated with Favorable Prognosis of IDH-WT Glioblastomas. Cancers 2019, 11, 1060. https://doi.org/10.3390/cancers11081060
Jovčevska I, Zottel A, Šamec N, Mlakar J, Sorokin M, Nikitin D, Buzdin AA, Komel R. High FREM2 Gene and Protein Expression Are Associated with Favorable Prognosis of IDH-WT Glioblastomas. Cancers. 2019; 11(8):1060. https://doi.org/10.3390/cancers11081060
Chicago/Turabian StyleJovčevska, Ivana, Alja Zottel, Neja Šamec, Jernej Mlakar, Maxim Sorokin, Daniil Nikitin, Anton A. Buzdin, and Radovan Komel. 2019. "High FREM2 Gene and Protein Expression Are Associated with Favorable Prognosis of IDH-WT Glioblastomas" Cancers 11, no. 8: 1060. https://doi.org/10.3390/cancers11081060
APA StyleJovčevska, I., Zottel, A., Šamec, N., Mlakar, J., Sorokin, M., Nikitin, D., Buzdin, A. A., & Komel, R. (2019). High FREM2 Gene and Protein Expression Are Associated with Favorable Prognosis of IDH-WT Glioblastomas. Cancers, 11(8), 1060. https://doi.org/10.3390/cancers11081060