Molecular Basis of Tumor Heterogeneity in Endometrial Carcinosarcoma

 , ,
, ,
Abstract
1. Clinicopathological Characteristics
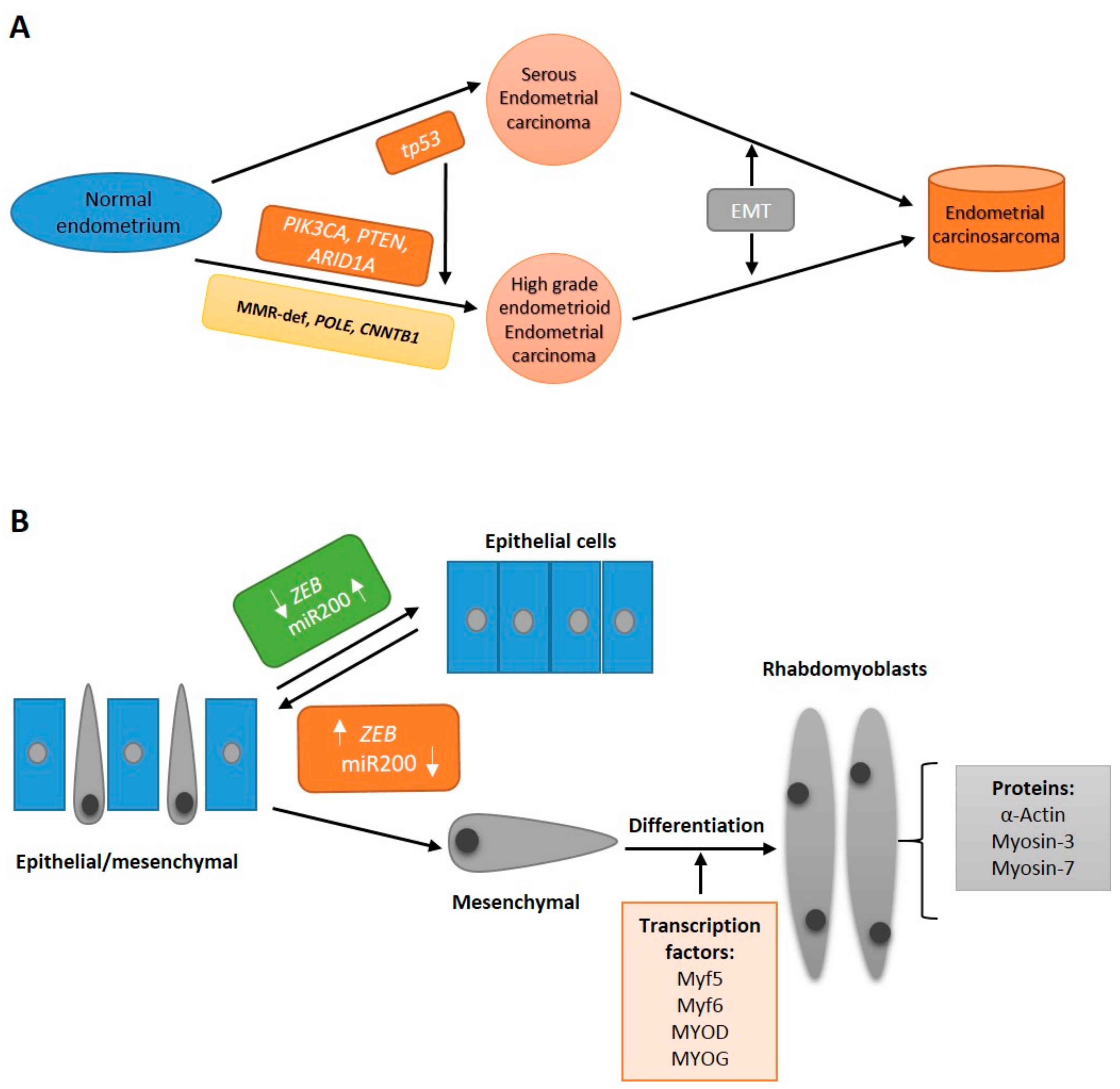
2. Molecular Subtypes of ECS
3. Serous-Like Molecular Alterations in ECS
4. Endometrioid-Like Molecular Alterations
5. Gene Expression Profiles in ECS
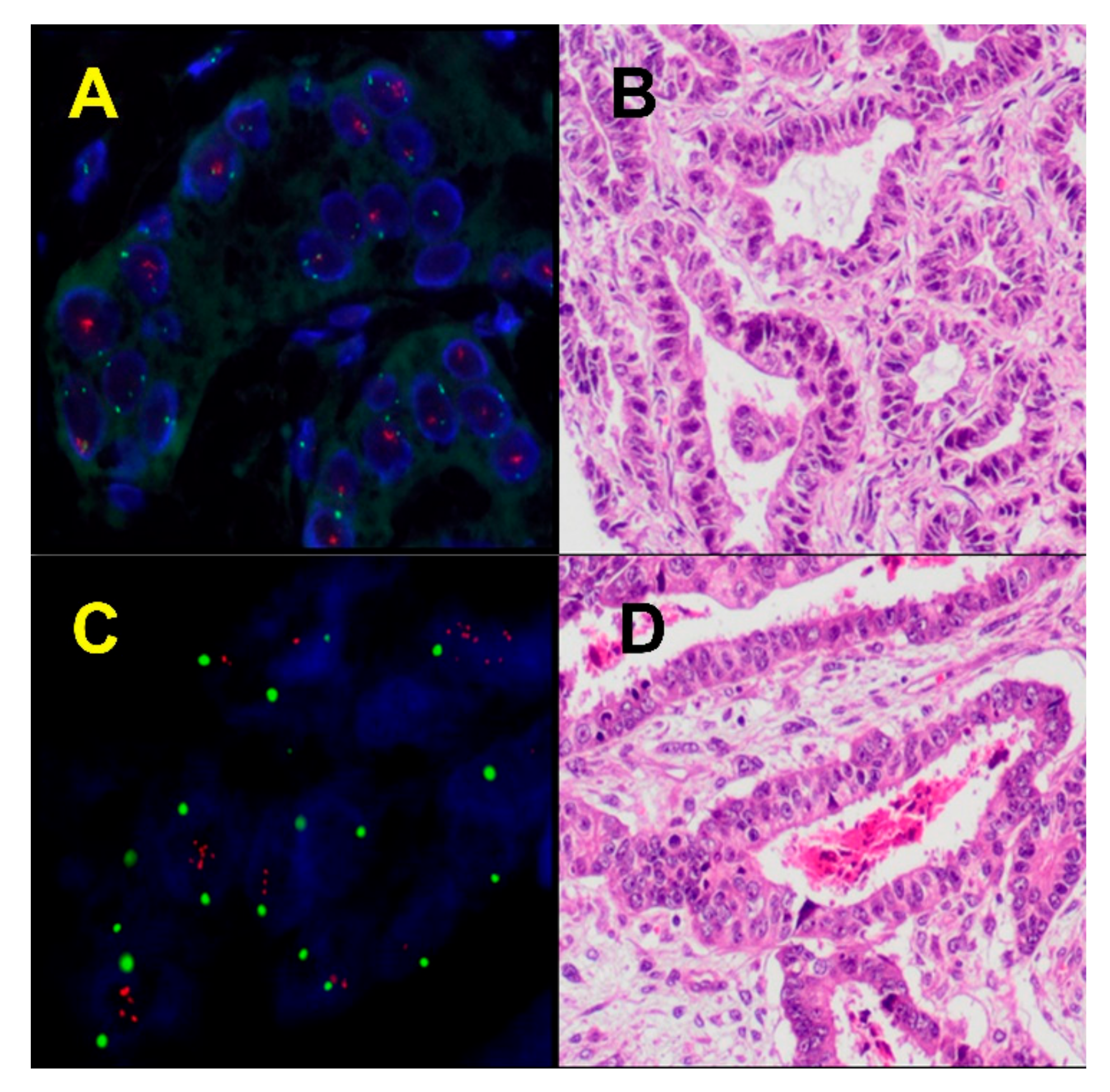
6. Methylation Profiles in ECS
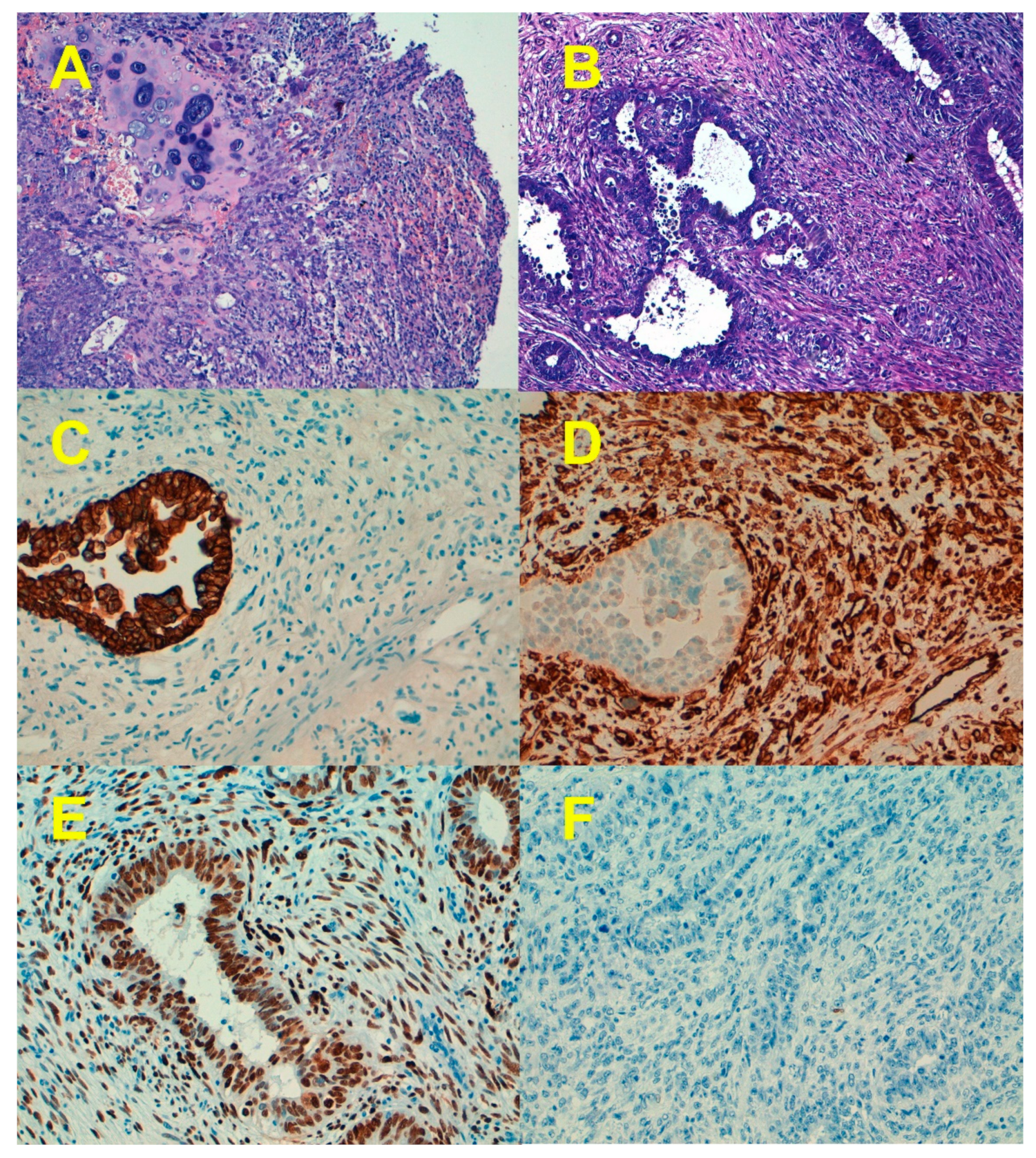
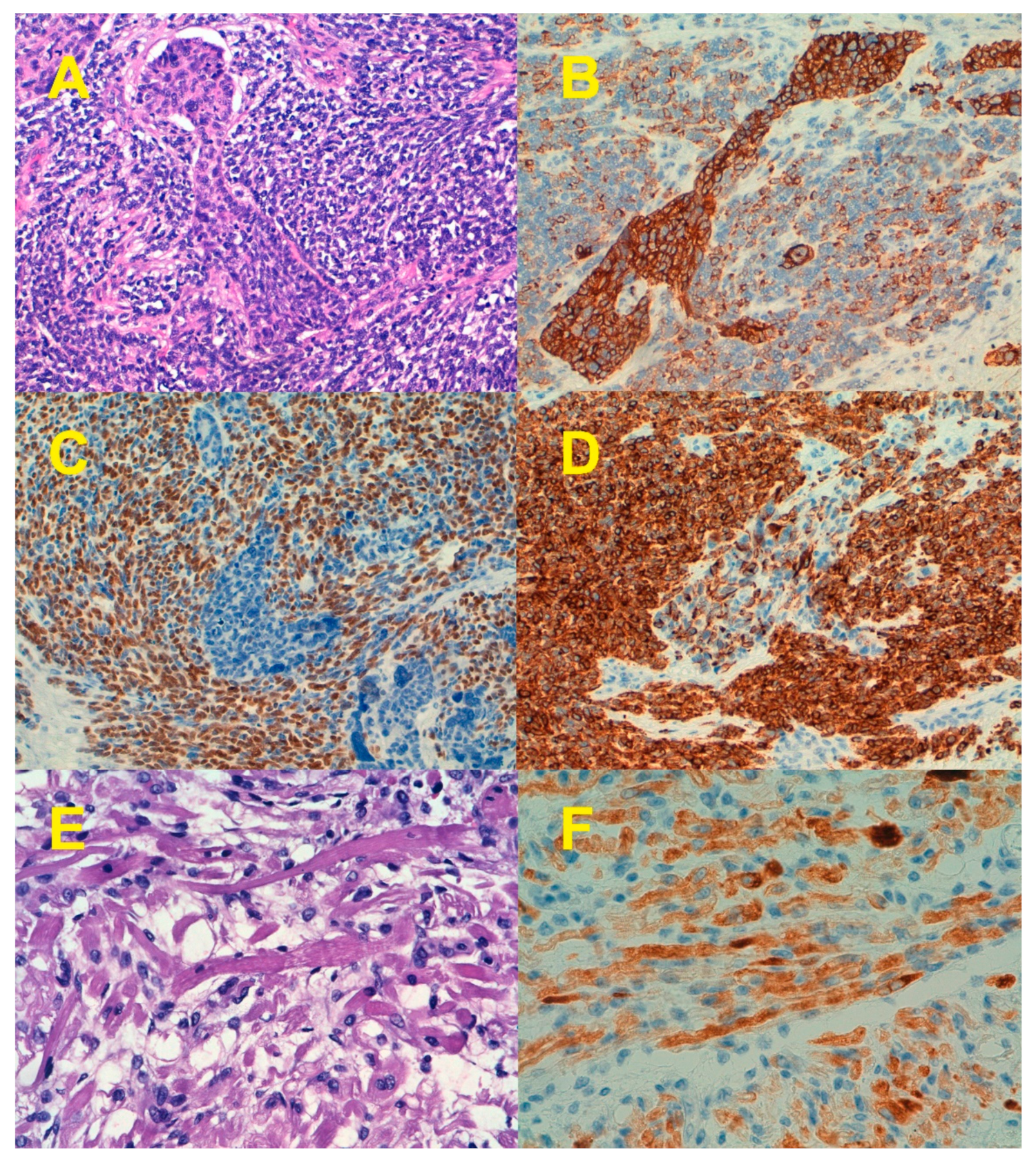
7. Epithelial-to-Mesenchymal Transition
8. Beyond EMT: Stemness and Differentiation in ECS
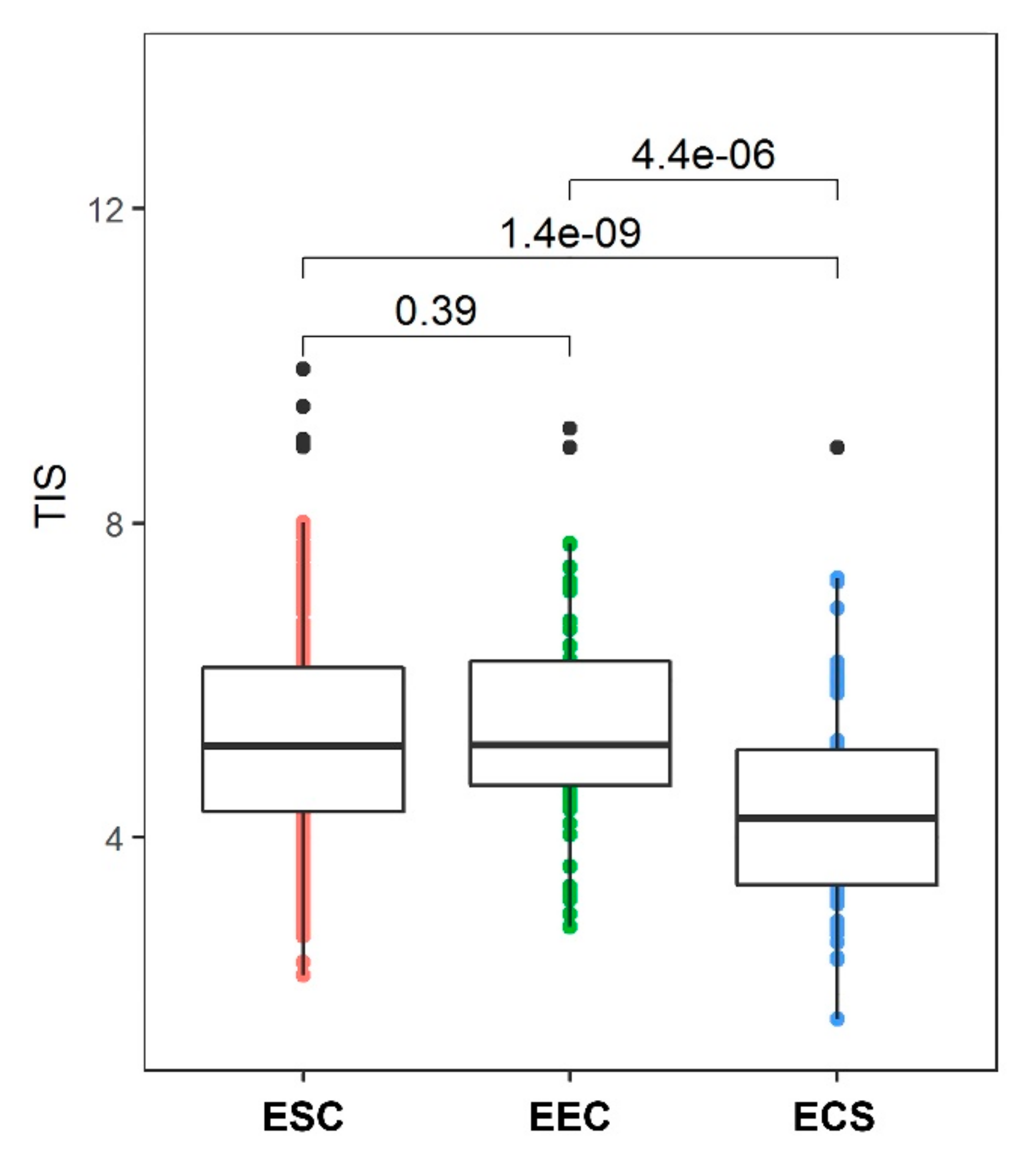
9. Immune Response in CS
10. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Tavassoli, F. World Health Organization Classification of Tumours. Pathology and Genetics of Tumors of Breast and Female Genital Organs; IARC Press: Lyon, France, 2003. [Google Scholar]
- Gonzalez Bosquet, J.; Terstriep, S.A.; Cliby, W.A.; Brown-Jones, M.; Kaur, J.S.; Podratz, K.C.; Keeney, G.L. The impact of multi-modal therapy on survival for uterine carcinosarcomas. Gynecol. Oncol. 2010, 116, 419–423. [Google Scholar] [CrossRef] [PubMed]
- De Jong, R.A.; Nijman, H.W.; Wijbrandi, T.F.; Reyners, A.K.; Boezen, H.M.; Hollema, H. Molecular markers and clinical behavior of uterine carcinosarcomas: Focus on the epithelial tumor component. Mod. Pathol. 2011, 24, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.E.; Tornos, C.; Hummer, A.; Barakat, R.R.; Soslow, R.A. Prognostic features of surgical stage I uterine carcinosarcoma. Am. J. Surg. Pathol. 2007, 31, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Amant, F.; Cadron, I.; Fuso, L.; Berteloot, P.; de Jonge, E.; Jacomen, G.; Van Robaeys, J.; Neven, P.; Moerman, P.; Vergote, I. Endometrial carcinosarcomas have a different prognosis and pattern of spread compared to high-risk epithelial endometrial cancer. Gynecol. Oncol. 2005, 98, 274–280. [Google Scholar] [CrossRef] [PubMed]
- George, E.; Lillemoe, T.J.; Twiggs, L.B.; Perrone, T. Malignant mixed mullerian tumor versus high-grade endometrial carcinoma and aggressive variants of endometrial carcinoma: A comparative analysis of survival. Int. J. Gynecol. Pathol. 1995, 14, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Callister, M.; Ramondetta, L.M.; Jhingran, A.; Burke, T.W.; Eifel, P.J. Malignant mixed mullerian tumors of the uterus: Analysis of patterns of failure, prognostic factors, and treatment outcome. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 786–796. [Google Scholar] [CrossRef]
- Bansal, N.; Herzog, T.J.; Seshan, V.E.; Schiff, P.B.; Burke, W.M.; Cohen, C.J.; Wright, J.D. Uterine carcinosarcomas and grade 3 endometrioid cancers: Evidence for distinct tumor behavior. Obstet. Gynecol. 2008, 112, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Ross, M.S.; Machida, H.; Blake, E.A.; Roman, L.D. Trends of uterine carcinosarcoma in the United States. J. Gynecol. Oncol. 2018, 29, e22. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.K.; Sung, C.O.; Kim, K.R. Uterine malignant mixed mullerian tumors following treatment with selective estrogen receptor modulators in patients with breast cancer: A report of 13 cases and their clinicopathologic characteristics. J. Pathol. Transl. Med. 2019, 53, 31–39. [Google Scholar] [CrossRef]
- Matsuo, K.; Ross, M.S.; Bush, S.H.; Yunokawa, M.; Blake, E.A.; Takano, T.; Ueda, Y.; Baba, T.; Satoh, S.; Shida, M.; et al. Tumor characteristics and survival outcomes of women with tamoxifen-related uterine carcinosarcoma. Gynecol. Oncol. 2017, 144, 329–335. [Google Scholar] [CrossRef]
- Buza, N.; Tavassoli, F.A. Comparative analysis of P16 and P53 expression in uterine malignant mixed mullerian tumors. Int. J. Gynecol. Pathol. 2009, 28, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, S.G.; Major, F.J.; Blessing, J.A.; Fetter, B.; Askin, F.B.; Liao, S.Y.; Miller, A. Carcinosarcoma (malignant mixed mesodermal tumor) of the uterus. A gynecologic oncology group pathologic study of 203 cases. Int. J. Gynecol. Pathol. 1990, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sreenan, J.J.; Hart, W.R. Carcinosarcomas of the female genital tract. A pathologic study of 29 metastatic tumors: Further evidence for the dominant role of the epithelial component and the conversion theory of histogenesis. Am. J. Surg. Pathol. 1995, 19, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.J.; Vogelsan, J.; Young, L.J. p53 gene mutation in female genital tract carcinosarcomas (malignant mixed mullerian tumors): A clinicopathologic study of 74 cases. Mod. Pathol. 1994, 7, 619–627. [Google Scholar] [PubMed]
- Matsuo, K.; Takazawa, Y.; Ross, M.S.; Elishaev, E.; Podzielinski, I.; Yunokawa, M.; Sheridan, T.B.; Bush, S.H.; Klobocista, M.M.; Blake, E.A.; et al. Significance of histologic pattern of carcinoma and sarcoma components on survival outcomes of uterine carcinosarcoma. Ann. Oncol. 2016, 27, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Abdulfatah, E.; Lordello, L.; Khurram, M.; Van de Vijver, K.; Alosh, B.; Bandyopadhyay, S.; Oliva, E.; Ali-Fehmi, R. Predictive histologic factors in carcinosarcomas of the uterus: A multiinstitutional study. Int. J. Gynecol. Pathol. 2018, 38, 205–215. [Google Scholar] [CrossRef]
- Costa, M.J.; Khan, R.; Judd, R. Carcinoma (malignant mixed mullerian [mesodermal] tumor) of the uterus and ovary. Correlation of clinical, pathologic, and immunohistochemical features in 29 cases. Arch. Pathol. Lab. Med. 1991, 115, 583–590. [Google Scholar] [PubMed]
- Kurnit, K.C.; Previs, R.A.; Soliman, P.T.; Westin, S.N.; Klopp, A.H.; Fellman, B.M.; Lu, K.H.; Ramondetta, L.M.; Fleming, N.D. Prognostic factors impacting survival in early stage uterine carcinosarcoma. Gynecol. Oncol. 2019, 152, 31–37. [Google Scholar] [CrossRef]
- McCluggage, W.G. A practical approach to the diagnosis of mixed epithelial and mesenchymal tumours of the uterus. Mod. Pathol. 2016, 29 (Suppl. 1), S78–S91. [Google Scholar] [CrossRef]
- Matsuo, K.; Takazawa, Y.; Ross, M.S.; Elishaev, E.; Yunokawa, M.; Sheridan, T.B.; Bush, S.H.; Klobocista, M.M.; Blake, E.A.; Takano, T.; et al. Significance of lymphovascular space invasion by the sarcomatous component in uterine carcinosarcoma. Ann. Surg. Oncol. 2018, 25, 2756–2766. [Google Scholar] [CrossRef]
- Iwasa, Y.; Haga, H.; Konishi, I.; Kobashi, Y.; Higuchi, K.; Katsuyama, E.; Minamiguchi, S.; Yamabe, H. Prognostic factors in uterine carcinosarcoma: A clinicopathologic study of 25 patients. Cancer 1998, 82, 512–519. [Google Scholar] [CrossRef]
- Matsuo, K.; Takazawa, Y.; Ross, M.S.; Elishaev, E.; Yunokawa, M.; Sheridan, T.B.; Bush, S.H.; Klobocista, M.M.; Blake, E.A.; Takano, T.; et al. Characterizing sarcoma dominance pattern in uterine carcinosarcoma: Homologous versus heterologous element. Surg. Oncol. 2018, 27, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Takazawa, Y.; Ross, M.S.; Elishaev, E.; Yunokawa, M.; Sheridan, T.B.; Bush, S.H.; Klobocista, M.M.; Blake, E.A.; Takano, T.; et al. Proposal for a risk-based categorization of uterine carcinosarcoma. Ann. Surg. Oncol. 2018, 25, 3676–3684. [Google Scholar] [CrossRef] [PubMed]
- Castilla, M.A.; Moreno-Bueno, G.; Romero-Perez, L.; Van De Vijver, K.; Biscuola, M.; López-García, M.Á.; Prat, J.; Matías-Guiu, X.; Cano, A.; Oliva, E.; et al. Micro-RNA signature of the epithelial-mesenchymal transition in endometrial carcinosarcoma. J. Pathol. 2011, 223, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Chiyoda, T.; Tsuda, H.; Tanaka, H.; Kataoka, F.; Nomura, H.; Nishimura, S.; Takano, M.; Susumu, N.; Saya, H.; Aoki, D. Expression profiles of carcinosarcoma of the uterine corpus-are these similar to carcinoma or sarcoma? Genes Chromosomes Cancer 2012, 51, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Wada, H.; Enomoto, T.; Fujita, M.; Yoshino, K.; Nakashima, R.; Kurachi, H.; Haba, T.; Wakasa, K.; Shroyer, K.R.; Tsujimoto, M.; et al. Molecular evidence that most but not all carcinosarcomas of the uterus are combination tumors. Cancer Res. 1997, 57, 5379–5385. [Google Scholar] [PubMed]
- Talhouk, A.; McConechy, M.K.; Leung, S.; Li-Chang, H.H.; Kwon, J.S.; Melnyk, N.; Yang, W.; Senz, J.; Boyd, N.; Karnezis, A.N.; et al. A clinically applicable molecular-based classification for endometrial cancers. Br. J. Cancer 2015, 113, 299–310. [Google Scholar] [CrossRef] [PubMed]
- McConechy, M.K.; Hoang, L.N.; Chui, M.H.; Senz, J.; Yang, W.; Rozenberg, N.; Mackenzie, R.; McAlpine, J.N.; Huntsman, D.G.; Clarke, B.A.; et al. In-depth molecular profiling of the biphasic components of uterine carcinosarcomas. J. Pathol. Clin. Res. 2015, 1, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.L.; Xiu, J.; Chatterjee-Paer, S.; Buckley de Meritens, A.; Burke, W.M.; Tergas, A.I.; Wright, J.D.; Hou, J.Y. Distinct molecular landscapes between endometrioid and nonendometrioid uterine carcinomas. Int. J. Cancer 2017, 140, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Rosa, J.M.; Leskela, S.; Cristobal-Lana, E.; Santón, A.; López-García, M.Á.; Muñoz, G.; Pérez-Mies, B.; Biscuola, M.; Prat, J.; Esther, O.E.; et al. Molecular genetic heterogeneity in undifferentiated endometrial carcinomas. Mod. Pathol. 2016, 29, 1390. [Google Scholar] [CrossRef] [PubMed]
- Kounelis, S.; Jones, M.W.; Papadaki, H.; Bakker, A.; Swalsky, P.; Finkelstein, S.D. Carcinosarcomas (malignant mixed mullerian tumors) of the female genital tract: Comparative molecular analysis of epithelial and mesenchymal components. Hum. Pathol. 1998, 29, 82–87. [Google Scholar] [CrossRef]
- Abeln, E.C.; Smit, V.T.; Wessels, J.W.; de Leeuw, W.J.; Cornelisse, C.J.; Fleuren, G.J. Molecular genetic evidence for the conversion hypothesis of the origin of malignant mixed mullerian tumours. J. Pathol. 1997, 183, 424–431. [Google Scholar] [CrossRef]
- Liu, F.S.; Kohler, M.F.; Marks, J.R.; Bast, R.C., Jr.; Boyd, J.; Berchuck, A. Mutation and overexpression of the p53 tumor suppressor gene frequently occurs in uterine and ovarian sarcomas. Obstet. Gynecol. 1994, 83, 118–124. [Google Scholar] [PubMed]
- Kanthan, R.; Senger, J.L.; Diudea, D. Malignant mixed Mullerian tumors of the uterus: Histopathological evaluation of cell cycle and apoptotic regulatory proteins. World J. Surg. Oncol. 2010, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Stransky, N.; McCord, C.L.; Cerami, E.; Lagowski, J.; Kelly, D.; Angiuoli, S.V.; Sausen, M.; Kann, L.; Shukla, M.; et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat. Commun. 2014, 5, 5006. [Google Scholar] [CrossRef] [PubMed]
- McConechy, M.K.; Ding, J.; Cheang, M.C.; Wiegand, K.; Senz, J.; Tone, A.; Yang, W.; Prentice, L.; Tse, K.; Zeng, T.; et al. Use of mutation profiles to refine the classification of endometrial carcinomas. J. Pathol. 2012, 228, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Cherniack, A.D.; Shen, H.; Walter, V.; Stewart, C.; Murray, B.A.; Bowlby, R.; Hu, X.; Ling, S.; Soslow, R.A.; Broaddus, R.R.; et al. Integrated molecular characterization of uterine carcinosarcoma. Cancer Cell 2017, 31, 411–423. [Google Scholar] [CrossRef]
- Liu, Y.; Weber, Z.; San Lucas, F.A.; Deshpande, A.; Jakubek, Y.A.; Sulaiman, R.; Fagerness, M.; Flier, N.; Sulaiman, J.; Davis, C.M.; et al. Assessing inter-component heterogeneity of biphasic uterine carcinosarcomas. Gynecol. Oncol. 2018, 151, 243–249. [Google Scholar] [CrossRef]
- Le Gallo, M.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program; Merino, M.J.; Mutch, D.G.; Goodfellow, P.J.; Mullikin, J.C.; Bell, D.W. The FOXA2 transcription factor is frequently somatically mutated in uterine carcinosarcomas and carcinomas. Cancer 2018, 124, 65–73. [Google Scholar] [CrossRef]
- D’Brot, A.; Kurtz, P.; Regan, E.; Jakubowski, B.; Abrams, J.M. A platform for interrogating cancer-associated p53 alleles. Oncogene 2017, 36, 286–291. [Google Scholar] [CrossRef]
- Semczuk, A.; Skomra, D.; Chyzynska, M.; Szewczuk, W.; Olcha, P.; Korobowicz, E. Immunohistochemical analysis of carcinomatous and sarcomatous components in the uterine carcinosarcoma: A case report. Pathol. Res. Pract. 2008, 204, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Reid-Nicholson, M.; Iyengar, P.; Hummer, A.J.; Linkov, I.; Asher, M.; Soslow, R.A. Immunophenotypic diversity of endometrial adenocarcinomas: Implications for differential diagnosis. Mod. Pathol. 2006, 19, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Bennett, B.; Belch, R.Z.; Han, A.C. Loss of p16 in recurrent malignant mixed mullerian tumors of the uterus. Int. J. Gynecol. Cancer 2006, 16, 1354–1357. [Google Scholar] [PubMed]
- Saule, C.; Mouret-Fourme, E.; Briaux, A.; Becette, V.; Rouzier, R.; Houdayer, C.; Stoppa-Lyonnet, D. Risk of serous endometrial carcinoma in women with pathogenic BRCA1/2 variant after risk-reducing salpingo-oophorectomy. J. Natl. Cancer Inst. 2018, 110, 213–215. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, M.M.; Mooyaart, A.L.; Vreeswijk, M.P.; de Kroon, C.D.; van Wezel, T.; van Asperen, C.J.; Smit, V.T.; Dekkers, O.M.; Bosse, T. Linking uterine serous carcinoma to BRCA1/2-associated cancer syndrome: A meta-analysis and case report. Eur. J. Cancer 2017, 72, 215–225. [Google Scholar] [CrossRef]
- Shu, C.A.; Pike, M.C.; Jotwani, A.R.; Friebel, T.M.; Soslow, R.A.; Levine, D.A.; Nathanson, K.L.; Konner, J.A.; Arnold, A.G.; Bogomolniy, F.; et al. Uterine cancer after risk-reducing salpingo-oophorectomy without hysterectomy in women with BRCA mutations. JAMA Oncol. 2016, 2, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, Y.; Saigo, P.E.; Federici, M.G.; Boyd, J. Carcinosarcoma of the ovary in a patient with a germline BRCA2 mutation: Evidence for monoclonal origin. Gynecol. Oncol. 2000, 76, 226–229. [Google Scholar] [CrossRef]
- Carnevali, I.W.; Cimetti, L.; Sahnane, N.; Libera, L.; Cavallero, A.; Formenti, G.; Riva, C.; Tibiletti, M.G. Two cases of carcinosarcomas of the ovary involved in hereditary cancer syndromes. Int. J. Gynecol. Pathol. 2017, 36, 64–70. [Google Scholar] [CrossRef]
- Ghilli, M.; Mariniello, D.M.; Fanelli, G.; Cascione, F.; Fontana, A.; Cristaudo, A.; Cilotti, A.; Caligo, A.M.; Manca, G.; Colizzi, L.; et al. Carcinosarcoma of the breast: An aggressive subtype of metaplastic cancer. Report of a rare case in a young BRCA-1 mutated woman. Clin. Breast Cancer 2017, 17, e31–e35. [Google Scholar] [CrossRef]
- Zhao, S.; Bellone, S.; Lopez, S.; Thakral, D.; Schwab, C.; English, D.P.; Black, J.; Cocco, E.; Choi, J.; Zammataro, L.; et al. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc. Natl. Acad. Sci USA 2016, 113, 12238–12243. [Google Scholar] [CrossRef]
- Schipf, A.; Mayr, D.; Kirchner, T.; Diebold, J. Molecular genetic aberrations of ovarian and uterine carcinosarcomas--a CGH and FISH study. Virchows Arch. 2008, 452, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Livasy, C.A.; Reading, F.C.; Moore, D.T.; Boggess, J.F.; Lininger, R.A. EGFR expression and HER2/neu overexpression/amplification in endometrial carcinosarcoma. Gynecol. Oncol. 2006, 100, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Raspollini, M.R.; Susini, T.; Amunni, G.; Paglierani, M.; Taddei, A.; Marchionni, M.; Scarselli, G.; Taddei, G.L. COX-2, c-KIT and HER-2/neu expression in uterine carcinosarcomas: Prognostic factors or potential markers for targeted therapies? Gynecol. Oncol. 2005, 96, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Tsuda, H.; Kimura, M.; Okamoto, S.; Kita, T.; Kasamatsu, T.; Yamada, T.; Kikuchi, Y.; Honjo, H.; Matsubara, O. Different expression patterns of KIT, EGFR, and HER-2 (c-erbB-2) oncoproteins between epithelial and mesenchymal components in uterine carcinosarcoma. Cancer Sci. 2003, 94, 986–991. [Google Scholar] [CrossRef]
- Nicoletti, R.; Lopez, S.; Bellone, S.; Cocco, E.; Schwab, C.L.; Black, J.D.; Centritto, F.; Zhu, L.; Bonazzoli, E.; Buza, N.; et al. T-DM1, a novel antibody-drug conjugate, is highly effective against uterine and ovarian carcinosarcomas overexpressing HER2. Clin. Exp. Metastasis 2015, 32, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Biscuola, M.; Van de Vijver, K.; Castilla, M.A.; Romero-Pérez, L.; López-García, M.Á.; Díaz-Martín, J.; Matias-Guiu, X.; Oliva, E.; Palacios Calvo, J. Oncogene alterations in endometrial carcinosarcomas. Hum. Pathol. 2013, 44, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Cimbaluk, D.; Rotmensch, J.; Scudiere, J.; Gown, A.; Bitterman, P. Uterine carcinosarcoma: Immunohistochemical studies on tissue microarrays with focus on potential therapeutic targets. Gynecol. Oncol. 2007, 105, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.P.; Douglas, W.; Ellenson, L.H. Molecular alterations of EGFR and PIK3CA in uterine serous carcinoma. Gynecol. Oncol. 2009, 113, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Garabedian, M.J.; Logan, S.K. URI1 amplification in uterine carcinosarcoma associates with chemo-resistance and poor prognosis. Am. J. Cancer Res. 2015, 5, 2320–2329. [Google Scholar] [PubMed]
- Chui, M.H.; Have, C.; Hoang, L.N.; Shaw, P.; Lee, C.H.; Clarke, B.A. Genomic profiling identifies GPC5 amplification in association with sarcomatous transformation in a subset of uterine carcinosarcomas. J. Pathol. Clin. Res. 2018, 4, 69–78. [Google Scholar] [CrossRef]
- Murray, S.; Linardou, H.; Mountzios, G.; Manoloukos, M.; Markaki, S.; Eleutherakis-Papaiakovou, E.; Dimopoulos, M.A.; Papadimitriou, C.A. Low frequency of somatic mutations in uterine sarcomas: A molecular analysis and review of the literature. Mutat. Res. 2010, 686, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Growdon, W.B.; Roussel, B.N.; Scialabba, V.L.; Foster, R.; Dias-Santagata, D.; Iafrate, A.J.; Ellisen, L.W.; Tambouret, R.H.; Rueda, B.R.; Borger, D.R. Tissue-specific signatures of activating PIK3CA and RAS mutations in carcinosarcomas of gynecologic origin. Gynecol. Oncol. 2011, 121, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Makker, V.; Recio, F.O.; Ma, L.; Matulonis, U.A.; Lauchle, J.O.; Parmar, H.; Gilbert, H.N.; Ware, J.A.; Zhu, R.; Lu, S.; et al. A multicenter, single-arm, open-label, phase 2 study of apitolisib (GDC-0980) for the treatment of recurrent or persistent endometrial carcinoma (MAGGIE study). Cancer 2016, 122, 3519–3528. [Google Scholar] [CrossRef] [PubMed]
- López-García, M.Á.; Begoña, V.; Castilla, M.A.; Romero-Pérez, L.; Díaz-Martín, J.; Biscuola, M.; Palacios, C.J. Genetics of endometrial carcinoma. Cancer Genomics 2013, 11, 51. [Google Scholar]
- Mills, A.M.; Liou, S.; Ford, J.M.; Berek, J.S.; Pai, R.K.; Longacre, T.A. Lynch syndrome screening should be considered for all patients with newly diagnosed endometrial cancer. Am. J. Surg. Pathol. 2014, 38, 1501–1509. [Google Scholar] [CrossRef]
- Taylor, N.P.; Zighelboim, I.; Huettner, P.C.; Powell, M.A.; Gibb, R.K.; Rader, J.S.; Mutch, D.G.; Edmonston, T.B.; Goodfellow, P.J. DNA mismatch repair and TP53 defects are early events in uterine carcinosarcoma tumorigenesis. Mod. Pathol. 2006, 19, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Hoang, L.N.; Ali, R.H.; Lau, S.; Gilks, C.B.; Lee, C.H. Immunohistochemical survey of mismatch repair protein expression in uterine sarcomas and carcinosarcomas. Int. J. Gynecol. Pathol. 2014, 33, 483–491. [Google Scholar] [CrossRef]
- Hembree, T.N.; Teer, J.K.; Hakam, A.; Chiappori, A.A. Genetic investigation of uterine carcinosarcoma: Case report and cohort analysis. Cancer Control 2016, 23, 61–66. [Google Scholar] [CrossRef]
- Bhangoo, M.S.; Boasberg, P.; Mehta, P.; Elvin, J.A.; Ali, S.M.; Wu, W.; Klempner, S.J. Tumor mutational burden guides therapy in a treatment refractory POLE-mutant uterine carcinosarcoma. Oncologist 2018, 23, 518–523. [Google Scholar] [CrossRef]
- Maxwell, G.L.; Chandramouli, G.V.; Dainty, L.; Litzi, T.J.; Berchuck, A.; Barrett, J.C.; Risinger, J.I. Microarray analysis of endometrial carcinomas and mixed mullerian tumors reveals distinct gene expression profiles associated with different histologic types of uterine cancer. Clin. Cancer Res. 2005, 11, 4056–4066. [Google Scholar] [CrossRef]
- Whitehurst, A.W. Cause and consequence of cancer/testis antigen activation in cancer. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Romero-Perez, L.; Castilla, M.A.; Lopez-Garcia, M.A.; Díaz-Martín, J.; Biscuola, M.; Ramiro-Fuentes, S.; Oliva, E.; Matias-Guiu, X.; Prat, J.; Cano, A.; et al. Molecular events in endometrial carcinosarcomas and the role of high mobility group AT-hook 2 in endometrial carcinogenesis. Hum. Pathol. 2013, 44, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Risinger, J.I.; Chandramouli, G.V.; Maxwell, G.L.; Custer, M.; Pack, S.; Loukinov, D.; Aprelikova, O.; Litzi, T.; Schrump, D.S.; Murphy, S.K.; et al. Global expression analysis of cancer/testis genes in uterine cancers reveals a high incidence of BORIS expression. Clin. Cancer Res. 2007, 13, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Resnick, M.B.; Sabo, E.; Kondratev, S.; Kerner, H.; Spagnoli, G.C.; Yakirevich, E. Cancer-testis antigen expression in uterine malignancies with an emphasis on carcinosarcomas and papillary serous carcinomas. Int. J. Cancer 2002, 101, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Roszik, J.; Wang, W.L.; Livingston, J.A.; Roland, C.L.; Ravi, V.; Yee, C.; Hwu, P.; Futreal, A.; Lazar, A.J.; Patel, S.R.; et al. Overexpressed PRAME is a potential immunotherapy target in sarcoma subtypes. Clin. Sarcoma Res. 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Ratner, E.S.; Tuck, D.; Richter, C.; Nallur, S.; Patel, R.M.; Schultz, V.; Hui, P.; Schwartz, P.E.; Rutherford, T.J.; Weidhaas, J.B. MicroRNA signatures differentiate uterine cancer tumor subtypes. Gynecol. Oncol. 2010, 118, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Li, B.; Yang, Z.; Fang, H.; Zhang, G.M.; Feng, Z.H.; Huang, B. Regulation of HIF-1alpha and VEGF by miR-20b tunes tumor cells to adapt to the alteration of oxygen concentration. PLoS ONE 2009, 4, e7629. [Google Scholar] [CrossRef] [PubMed]
- Hovey, A.M.; Devor, E.J.; Breheny, P.J.; Mott, S.L.; Dai, D.; Thiel, K.W.; Leslie, K.K. miR-888: A novel cancer-testis antigen that targets the progesterone receptor in endometrial cancer. Transl. Oncol. 2015, 8, 85–96. [Google Scholar] [CrossRef][Green Version]
- Gonzalez Dos Anjos, L.; de Almeida, B.C.; Gomes de Almeida, T.; Mourão Lavorato Rocha, A.; De Nardo Maffazioli, G.; Soares, F.A.; Werneck da Cunha, I.; Chada Baracat, E.; Candido Carvalho, K. Could miRNA signatures be useful for predicting uterine sarcoma and carcinosarcoma prognosis and treatment? Cancers 2018, 10, 315. [Google Scholar] [CrossRef]
- Li, J.; Xing, X.; Li, D.; Zhang, B.; Mutch, D.G.; Hagemann, I.S.; Wang, T. Whole-genome DNA methylation profiling identifies epigenetic signatures of uterine carcinosarcoma. Neoplasia 2017, 19, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Martin, J.; Diaz-Lopez, A.; Moreno-Bueno, G.; Castilla, M.Á.; Rosa-Rosa, J.M.; Cano, A.; Palacios, J. A core microRNA signature associated with inducers of the epithelial-to-mesenchymal transition. J. Pathol. 2014, 232, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Lopez, A.; Diaz-Martin, J.; Moreno-Bueno, G.; Cuevas, E.P.; Santos, V.; Olmeda, D.; Portillo, F.; Palacios, J.; Cano, A. Zeb1 and Snail1 engage miR-200f transcriptional and epigenetic regulation during EMT. Int. J. Cancer 2015, 136, E62–E73. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 2018, 33, 690–705. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Hashimura, M.; Akiya, M.; Chiba, R.; Saegusa, M. Functional role of ALK-related signal cascades on modulation of epithelial-mesenchymal transition and apoptosis in uterine carcinosarcoma. Mol. Cancer 2017, 16, 37. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gao, F.; Liu, N. L1CAM promotes epithelial to mesenchymal transition and formation of cancer initiating cells in human endometrial cancer. Exp. Ther. Med. 2018, 15, 2792–2797. [Google Scholar] [CrossRef]
- Versluis, M.; Plat, A.; de Bruyn, M.; Matias-Guiu, X.; Trovic, J.; Krakstad, C.; Nijman, H.W.; Bosse, T.; de Bock, G.H.; Hollema, H. L1CAM expression in uterine carcinosarcoma is limited to the epithelial component and may be involved in epithelial-mesenchymal transition. Virchows Arch. 2018, 473, 591–598. [Google Scholar] [CrossRef]
- Jolly, M.K.; Jia, D.; Boareto, M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Coupling the modules of EMT and stemness: A tunable ‘stemness window’ model. Oncotarget 2015, 6, 25161–25174. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Wang, H.; Unternaehrer, J.J. Epithelial-mesenchymal transition and cancer stem cells: At the crossroads of differentiation and dedifferentiation. Dev. Dyn. 2019, 248, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhang, L.; Zhao, H.; Chen, C.; Wang, Y.; Liu, S.; Lin, X.; Wang, Y.; Zhang, Q.; Lu, T.; et al. Molecular classification and subtype-specific drug sensitivity research of uterine carcinosarcoma under multi-omics framework. Cancer Biol. Ther. 2019, 20, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Longoria, T.C.; Eskander, R.N. Immunotherapy in endometrial cancer–an evolving therapeutic paradigm. Gynecol. Oncol. Res. Pract. 2015, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Danaher, P.; Warren, S.; Lu, R.; Samayoa, J.; Sullivan, A.; Pekker, I.; Wallden, B.; Marincola, F.M.; Cesano, A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): Results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 2018, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef]
- Ueno, T.; Tsuchikawa, T.; Hatanaka, K.C.; Hatanaka, Y.; Mitsuhashi, T.; Nakanishi, Y.; Noji, T.; Nakamura, T.; Okamura, K.; Matsuno, Y.; et al. Prognostic impact of programmed cell death ligand 1 (PD-L1) expression and its association with epithelial-mesenchymal transition in extrahepatic cholangiocarcinoma. Oncotarget 2018, 9, 20034–20047. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef]
- Kim, S.; Koh, J.; Kim, M.Y.; Kwon, D.; Go, H.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H. PD-L1 expression is associated with epithelial-to-mesenchymal transition in adenocarcinoma of the lung. Hum. Pathol. 2016, 58, 7–14. [Google Scholar] [CrossRef]
- Ock, C.Y.; Kim, S.; Keam, B.; Kim, M.; Kim, T.M.; Kim, J.H.; Jeon, Y.K.; Lee, J.S.; Kwon, S.K.; Hah, J.H.; et al. PD-L1 expression is associated with epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 15901–15914. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A patient-derived, pan-cancer EMT signature identifies global Molecular alterations and immune target enrichment following epithelial-to-mesenchymal transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: Beyond immune evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef] [PubMed]
- Velcheti, V.; Rimm, D.L.; Schalper, K.A. Sarcomatoid lung carcinomas show high levels of programmed death ligand-1 (PD-L1). J. Thorac. Oncol. 2013, 8, 803–805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, Y.; Ma, M.; Hao, J.; Ding, R.; Han, L.; Zou, J.; Zhang, L.; Meng, Q.; Qu, X.; et al. Significant benefit of Nivolumab treating PD-L1 positive metastatic pulmonary carcinosarcoma: A case report and literature review. Oncotarget 2017, 8, 96453–96459. [Google Scholar] [CrossRef]
- Zhu, J.; Wen, H.; Ju, X.; Bi, R.; Zuo, W.; Wu, X. Clinical significance of programmed death ligand1 and intra-tumoral CD8+ T lymphocytes in ovarian carcinosarcoma. PLoS ONE 2017, 12, e0170879. [Google Scholar]
- Pinto, A.; Mackrides, N.; Nadji, M. PD-L1 expression in carcinosarcomas of the gynecologic tract: A potentially actionable biomarker. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 393–397. [Google Scholar] [CrossRef]
- Remmerie, M.; Janssens, V. Targeted therapies in type II endometrial cancers: Too little, but not too late. Int. J. Mol. Sci. 2018, 19, 2380. [Google Scholar] [CrossRef]
- Clinical Trials. Available online: clinicaltrials.gov (accessed on 26 June 2019).
- Fader, A.N.; Roque, D.M.; Siegel, E.; Buza, N.; Hui, P.; Abdelghany, O.; Chambers, S.K.; Secord, A.A.; Havrilesky, L.; O’Malley, D.M.; et al. Randomized phase II trial of carboplatin-paclitaxel versus carboplatin-paclitaxel-trastuzumab in uterine serous carcinomas that overexpress human epidermal growth factor receptor 2/neu. J. Clin. Oncol. 2018, 36, 2044–2051. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | Endometrioid Carcinoma | Serous Carcinoma | Carcinosarcoma |
|---|---|---|---|
| PTEN | 82% | 10% | 19% |
| PIK3CA | 54% | 37% | 35% |
| PIK3R1 | 36% | 11% | 11% |
| CTNNB1 | 34% | 1% | 2% |
| ARID1A | 54% | 8% | 12% |
| KRAS | 24% | 3% | 12% |
| CTCF | 31% | 2% | 7% |
| TP53 | 21% | 88% | 91% |
| FBXW7 | 17% | 24% | 39% |
| PPP2R1A | 11% | 38% | 28% |
| CHD4 | 9% | 18% | 17% |
| CCNE1 (ampl.) | 16% | 26% | 41% |
| MYC (ampl.) | 14% | 24% | 21% |
| MECOM (ampl.) | 18% | 33% | 18% |
| PIK3CA (ampl.) | 10% | 22% | 11% |
| ERBB2 (ampl.) | 8% | 19% | 9% |
| Gene | Cherniack (n = 57) | McConechy (n = 30) | Jones (n = 361) | Zhao (n = 64) * | Le Gallo (n = 53) |
|---|---|---|---|---|---|
| TP53 | 91% | 80% | 67% | ~80% | 76% |
| FBXW7 | 39% | 20% | ~22% | 19% | |
| PIK3CA | 35% | 40% | 22% | ~20% | 34% |
| PPP2R1A | 28% | 13% | ~25% | 19% | |
| PTEN | 19% | 27% | ~7% | ||
| CHD4 | 17% | ~20% | 17% | ||
| ARID1A | 12% | 10% | ~4% | ||
| KRAS | 12% | 10% | ~4% | ||
| PIK3R1 | 11% | 17% | ~4% | ||
| AKT3 | 5% | ||||
| BRCA1 | 18% | ||||
| BRCA2 | 27% | ||||
| ZFHX3 | 7% | ||||
| CSMD3 | 23% | ||||
| HIST1H2BJ/G | 21% | ||||
| FOXA2 | 15% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leskela, S.; Pérez-Mies, B.; Rosa-Rosa, J.M.; Cristobal, E.; Biscuola, M.; Palacios-Berraquero, M.L.; Ong, S.; Matias-Guiu Guia, X.; Palacios, J. Molecular Basis of Tumor Heterogeneity in Endometrial Carcinosarcoma. Cancers 2019, 11, 964. https://doi.org/10.3390/cancers11070964
Leskela S, Pérez-Mies B, Rosa-Rosa JM, Cristobal E, Biscuola M, Palacios-Berraquero ML, Ong S, Matias-Guiu Guia X, Palacios J. Molecular Basis of Tumor Heterogeneity in Endometrial Carcinosarcoma. Cancers. 2019; 11(7):964. https://doi.org/10.3390/cancers11070964
Chicago/Turabian StyleLeskela, Susanna, Belen Pérez-Mies, Juan Manuel Rosa-Rosa, Eva Cristobal, Michele Biscuola, María L. Palacios-Berraquero, SuFey Ong, Xavier Matias-Guiu Guia, and José Palacios. 2019. "Molecular Basis of Tumor Heterogeneity in Endometrial Carcinosarcoma" Cancers 11, no. 7: 964. https://doi.org/10.3390/cancers11070964
APA StyleLeskela, S., Pérez-Mies, B., Rosa-Rosa, J. M., Cristobal, E., Biscuola, M., Palacios-Berraquero, M. L., Ong, S., Matias-Guiu Guia, X., & Palacios, J. (2019). Molecular Basis of Tumor Heterogeneity in Endometrial Carcinosarcoma. Cancers, 11(7), 964. https://doi.org/10.3390/cancers11070964