Combined Treatment with MEK and mTOR Inhibitors is Effective in In Vitro and In Vivo Models of Hepatocellular Carcinoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
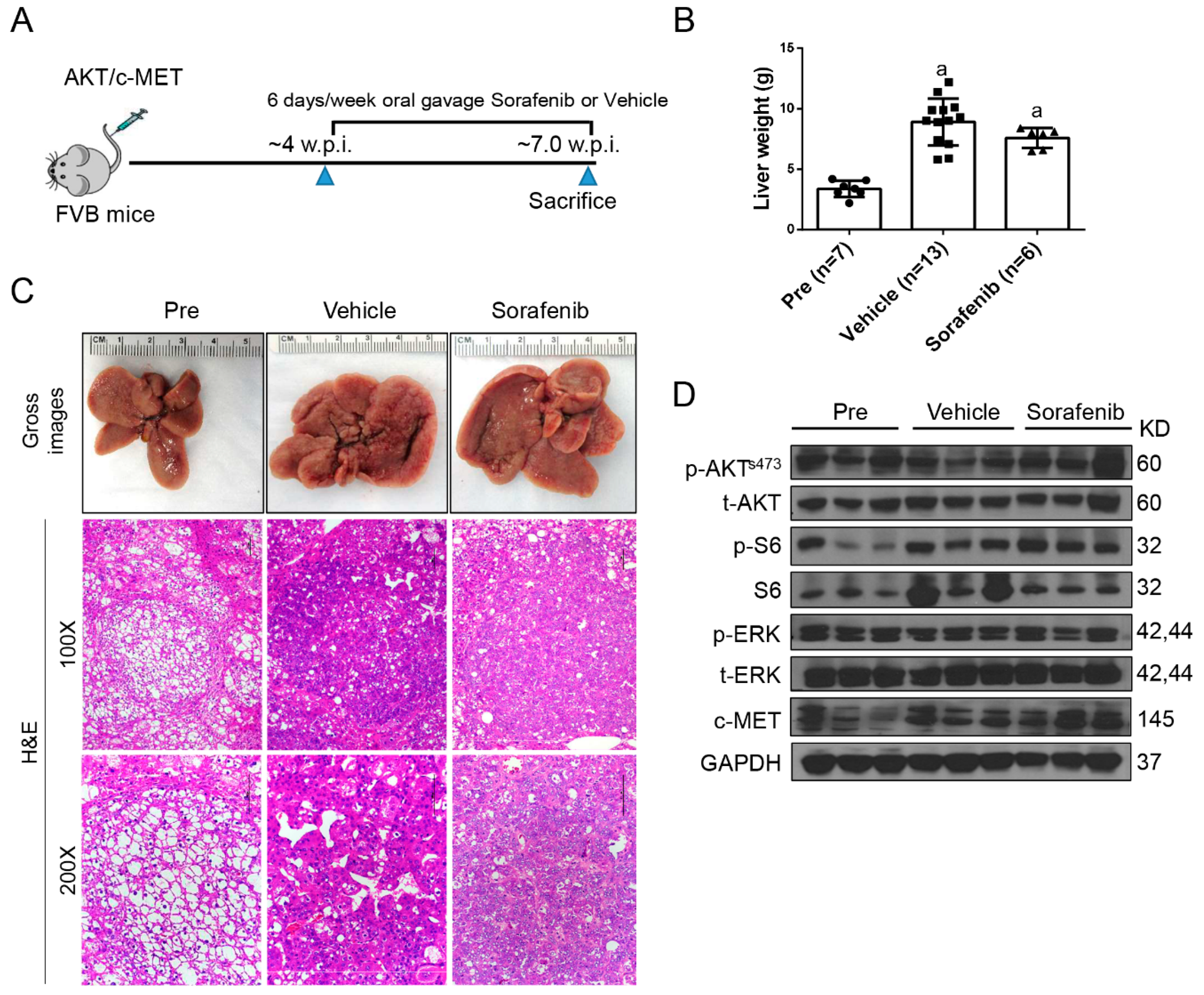
2.1. Limited Efficacy of Sorafenib and Regorafenib in HCC from AKT/c-MET Mice
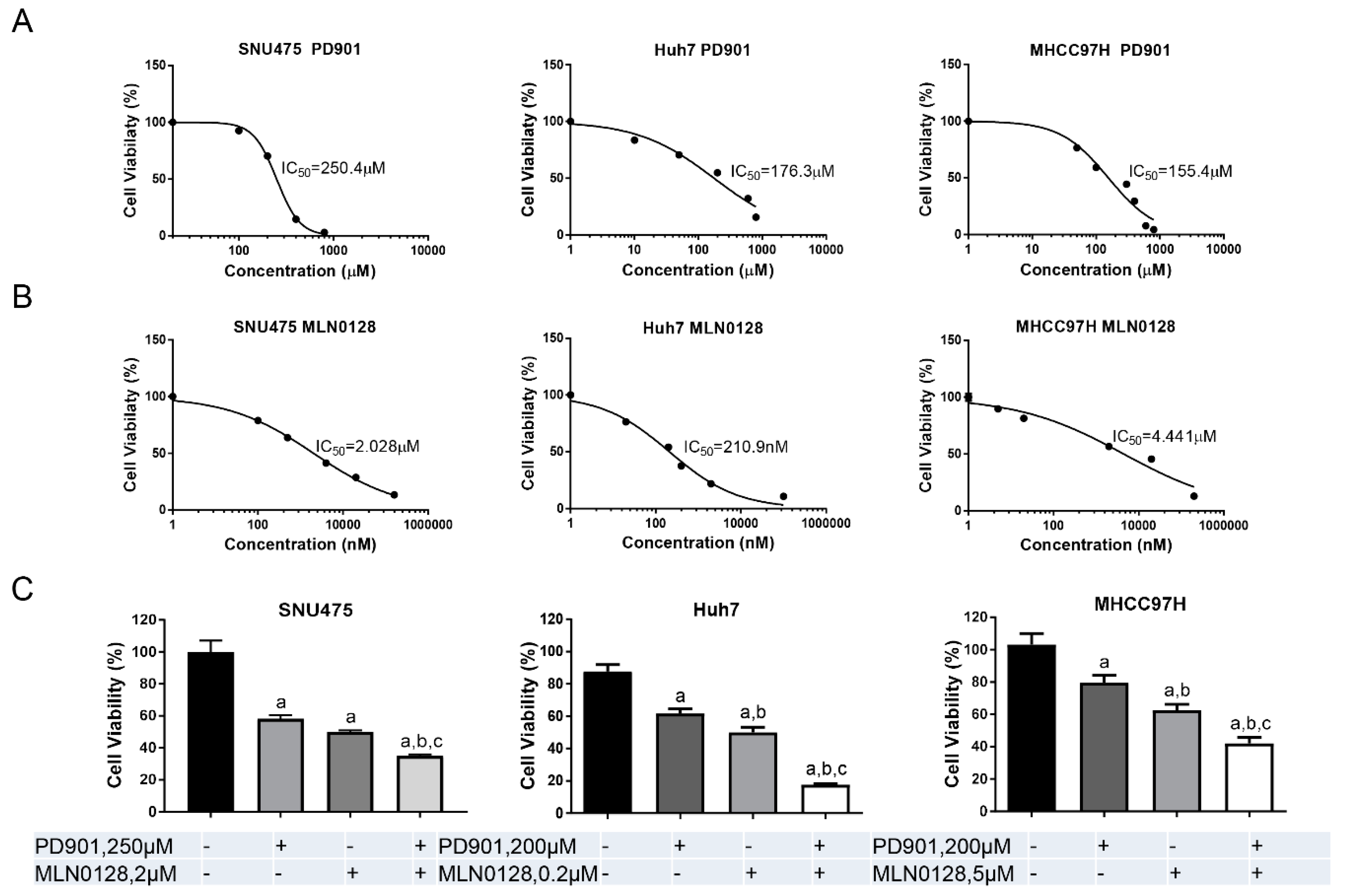
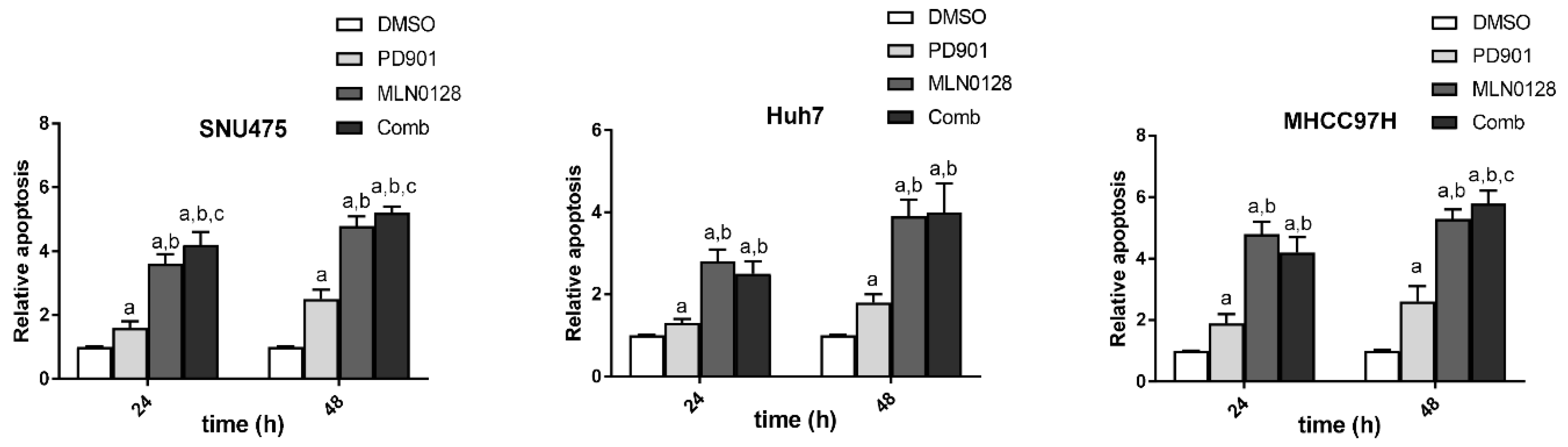
2.2. Increased Growth Inhibition in Human HCC Cell Lines by PD901 and MLN0128
2.3. PD901 and MLN0128 Combination Therapy Results in a Stable Disease in AKT/c-MET Mice
2.4. Combined PD901/MLN0128 Regimen Inhibits Tumor Cell Proliferation In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Hydrodynamic Tail Vein Injection and Mouse Treatment
4.3. Histology and Immunohistochemistry
4.4. Western Blot Analysis
4.5. In Vitro Experiments
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arslanoglu, A.; Seyal, A.R.; Sodagari, F.; Sahin, A.; Miller, F.H.; Salem, R.; Yaghmai, V. Current Guidelines for the Diagnosis and Management of Hepatocellular Carcinoma: A Comparative Review. Am. J. Roentgenol. 2016, 207, W88–W98. [Google Scholar] [CrossRef] [PubMed]
- Pascual, S.; Herrera, I.; Irurzun, J. New advances in hepatocellular carcinoma. World J. Hepatol. 2016, 8, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Liccioni, A.; Reig, M.; Bruix, J. Treatment of hepatocellular carcinoma. Dig. Dis. 2014, 32, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Waidmann, O. Recent developments with immunotherapy for hepatocellular carcinoma. Expert Opin. Biol. Ther. 2018, 18, 905–910. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Ciruelos Gil, E.M. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat. Rev. 2014, 40, 862–871. [Google Scholar] [CrossRef]
- Polivka, J., Jr.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.J.; Lang, H.; Wohlschlaeger, J.; Sotiropoulos, G.C.; Reis, H.; Schmid, K.W.; Baba, H.A. AKT and ERK1/2 signaling in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2007, 13, 6470–6477. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008, 135, 1972–1983.e11. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Siegel, D.S.; Vij, R.; Berdeja, J.G.; Richardson, P.G.; Neuwirth, R.; Patel, C.G.; Zohren, F.; Wolf, J.L. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: A phase I dose escalation study in patients with relapsed or refractory multiple myeloma, non-Hodgkin lymphoma, or Waldenstrom’s macroglobulinemia. Am. J. Hematol. 2016, 91, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Badawi, M.; Kim, J.; Dauki, A.; Sutaria, D.; Motiwala, T.; Reyes, R.; Wani, N.; Kolli, S.; Jiang, J.; Coss, C.C.; et al. CD44 positive and sorafenib insensitive hepatocellular carcinomas respond to the ATP-competitive mTOR inhibitor INK128. Oncotarget 2018, 9, 26032–26045. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; de Mestier, L.; Cros, J.; Faivre, S.; Raymond, E. MEK in cancer and cancer therapy. Pharmacol. Ther. 2014, 141, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Hoffmann, K.; Xiao, Z.; Jin, N.; Galli, U.; Mohr, E.; Buchler, M.W.; Schemmer, P. MEK inhibition induced downregulation of MRP1 and MRP3 expression in experimental hepatocellular carcinoma. Cancer Cell Int. 2013, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Che, L.; Li, L.; Pilo, M.G.; Cigliano, A.; Ribback, S.; Li, X.; Latte, G.; Mela, M.; Evert, M.; et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci. Rep. 2016, 6, 20484. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Zhang, C.; Zhou, J.; Hu, J.; Sheng, L.; Li, X.; Chen, L.; Li, X.; Deng, X.; Zheng, G. Celecoxib alleviates AKT/c-Met-triggered rapid hepatocarcinogenesis by suppressing a novel COX-2/AKT/FASN cascade. Mol. Carcinog. 2019, 58, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hu, J.; Sheng, L.; Yuan, M.; Wu, Y.; Chen, L.; Zheng, G.; Qiu, Z. Metformin delays AKT/c-Met-driven hepatocarcinogenesis by regulating signaling pathways for de novo lipogenesis and ATP generation. Toxicol. Appl. Pharmacol. 2019, 365, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef]
- Dong, M.; Liu, X.; Evert, K.; Utpatel, K.; Peters, M.; Zhang, S.; Xu, Z.; Che, L.; Cigliano, A.; Ribback, S.; et al. Efficacy of MEK inhibition in a K-Ras-driven cholangiocarcinoma preclinical model. Cell Death Dis. 2018, 9, 31. [Google Scholar] [CrossRef]
- Zhang, S.; Song, X.; Cao, D.; Xu, Z.; Fan, B.; Che, L.; Hu, J.; Chen, B.; Dong, M.; Pilo, M.G.; et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J. Hepatol. 2017, 67, 1194–1203. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef]
- Yu, X.N.; Chen, H.; Liu, T.T.; Wu, J.; Zhu, J.M.; Shen, X.Z. Targeting the mTOR regulatory network in hepatocellular carcinoma: Are we making headway? Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Buontempo, F.; McCubrey, J.A. Drug discovery targeting the mTOR pathway. Clin. Sci. (Lond.) 2018, 132, 543–568. [Google Scholar] [CrossRef] [PubMed]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the mTOR pathway in hepatocellular carcinoma: Current state and future trends. J. Hepatol. 2014, 60, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Duan, Y.; Zheng, X.F. Targeting the mTOR kinase domain: The second generation of mTOR inhibitors. Drug Discov. Today 2011, 16, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Grant, S. Cotargeting survival signaling pathways in cancer. J. Clin. Investig. 2008, 118, 3003–3006. [Google Scholar] [CrossRef] [PubMed]
- Yuen, H.F.; Abramczyk, O.; Montgomery, G.; Chan, K.K.; Huang, Y.H.; Sasazuki, T.; Shirasawa, S.; Gopesh, S.; Chan, K.W.; Fennell, D.; et al. Impact of oncogenic driver mutations on feedback between the PI3K and MEK pathways in cancer cells. Biosci. Rep. 2012, 32, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; Troiani, T.; D’Aiuto, E.; Morgillo, F.; Vitagliano, D.; Capasso, A.; Costantino, S.; Ciuffreda, L.P.; Merolla, F.; Vecchione, L.; et al. Antitumor activity of pimasertib, a selective MEK 1/2 inhibitor, in combination with PI3K/mTOR inhibitors or with multi-targeted kinase inhibitors in pimasertib-resistant human lung and colorectal cancer cells. Int. J. Cancer 2013, 133, 2089–2101. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.R.; Michael, H.T.; Halsey, C.H.; Peer, C.J.; Adhikari, A.; Dwyer, J.E.; Hoover, S.B.; El Meskini, R.; Kozlov, S.; Weaver Ohler, Z.; et al. Synergistic targeted inhibition of MEK and dual PI3K/mTOR diminishes viability and inhibits tumor growth of canine melanoma underscoring its utility as a preclinical model for human mucosal melanoma. Pigment. Cell Melanoma Res. 2016, 29, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Hikita, H.; Takehara, T.; Shimizu, S.; Kodama, T.; Shigekawa, M.; Iwase, K.; Hosui, A.; Miyagi, T.; Tatsumi, T.; Ishida, H.; et al. The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology 2010, 52, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Tutusaus, A.; Stefanovic, M.; Boix, L.; Cucarull, B.; Zamora, A.; Blasco, L.; de Frutos, P.G.; Reig, M.; Fernandez-Checa, J.C.; Mari, M.; et al. Antiapoptotic BCL-2 proteins determine sorafenib/regorafenib resistance and BH3-mimetic efficacy in hepatocellular carcinoma. Oncotarget 2018, 9, 16701–16717. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Marti, P.; Stein, C.; Blumer, T.; Abraham, Y.; Dill, M.T.; Pikiolek, M.; Orsini, V.; Jurisic, G.; Megel, P.; Makowska, Z.; et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology 2015, 62, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Calvisi, D.F.; Ranganathan, S.; Cigliano, A.; Zhou, L.; Singh, S.; Jiang, L.; Fan, B.; Terracciano, L.; Armeanu-Ebinger, S.; et al. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 2014, 147, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Calvisi, D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Hu, J.; Song, X.; Utpatel, K.; Zhang, Y.; Wang, P.; Lu, X.; Zhang, J.; Xu, M.; Su, T.; et al. Combined Treatment with MEK and mTOR Inhibitors is Effective in In Vitro and In Vivo Models of Hepatocellular Carcinoma. Cancers 2019, 11, 930. https://doi.org/10.3390/cancers11070930
Liu X, Hu J, Song X, Utpatel K, Zhang Y, Wang P, Lu X, Zhang J, Xu M, Su T, et al. Combined Treatment with MEK and mTOR Inhibitors is Effective in In Vitro and In Vivo Models of Hepatocellular Carcinoma. Cancers. 2019; 11(7):930. https://doi.org/10.3390/cancers11070930
Chicago/Turabian StyleLiu, Xianqiong, Junjie Hu, Xinhua Song, Kirsten Utpatel, Yi Zhang, Pan Wang, Xinjun Lu, Jie Zhang, Meng Xu, Tao Su, and et al. 2019. "Combined Treatment with MEK and mTOR Inhibitors is Effective in In Vitro and In Vivo Models of Hepatocellular Carcinoma" Cancers 11, no. 7: 930. https://doi.org/10.3390/cancers11070930
APA StyleLiu, X., Hu, J., Song, X., Utpatel, K., Zhang, Y., Wang, P., Lu, X., Zhang, J., Xu, M., Su, T., Che, L., Wang, J., Evert, M., Calvisi, D. F., & Chen, X. (2019). Combined Treatment with MEK and mTOR Inhibitors is Effective in In Vitro and In Vivo Models of Hepatocellular Carcinoma. Cancers, 11(7), 930. https://doi.org/10.3390/cancers11070930