DNA-Repair Gene Mutations Are Highly Prevalent in Circulating Tumour DNA from Multiple Myeloma Patients

 ,
,  and
and 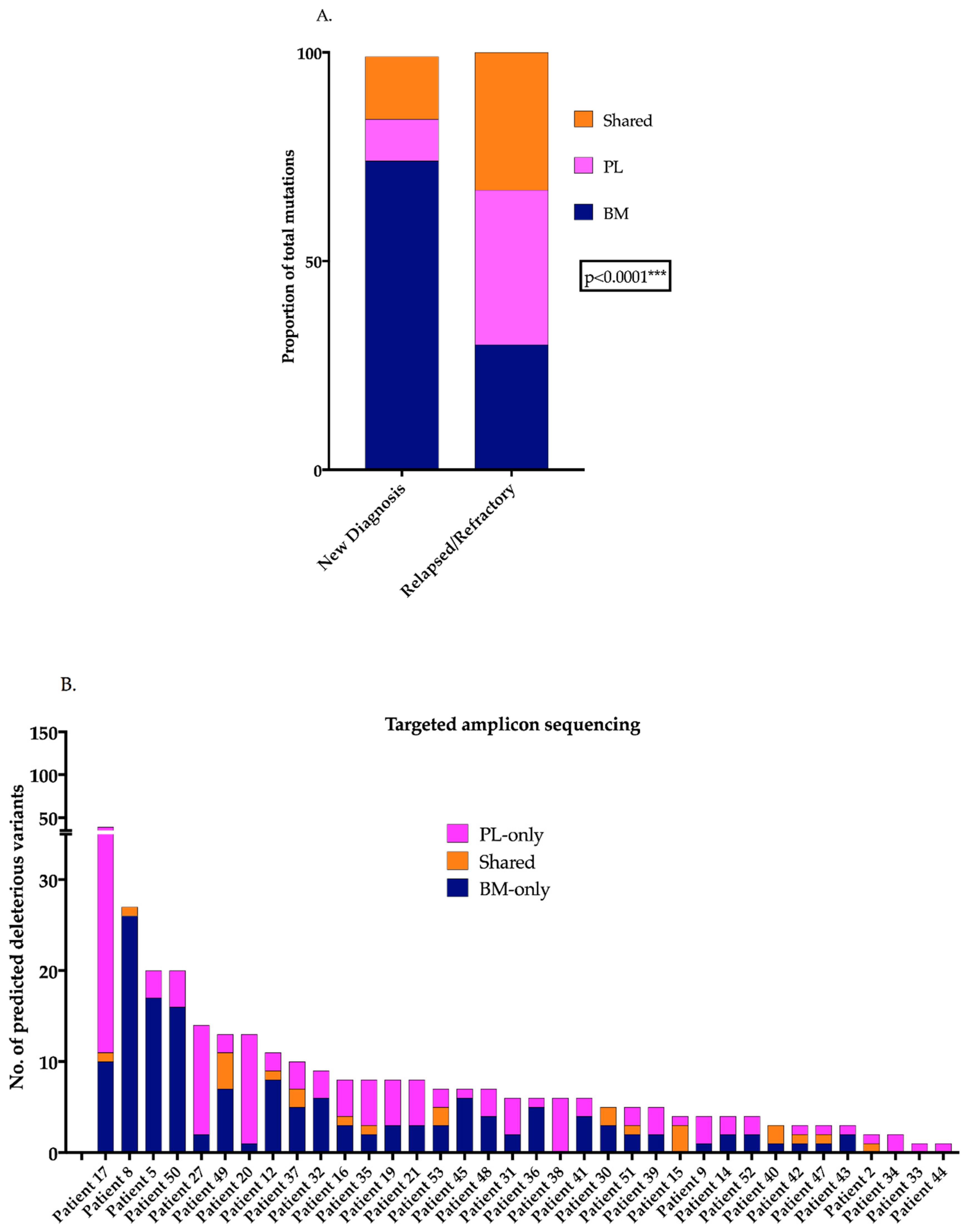{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. PL-Specific Mutations Characterise the Advanced MM Tumour Genome
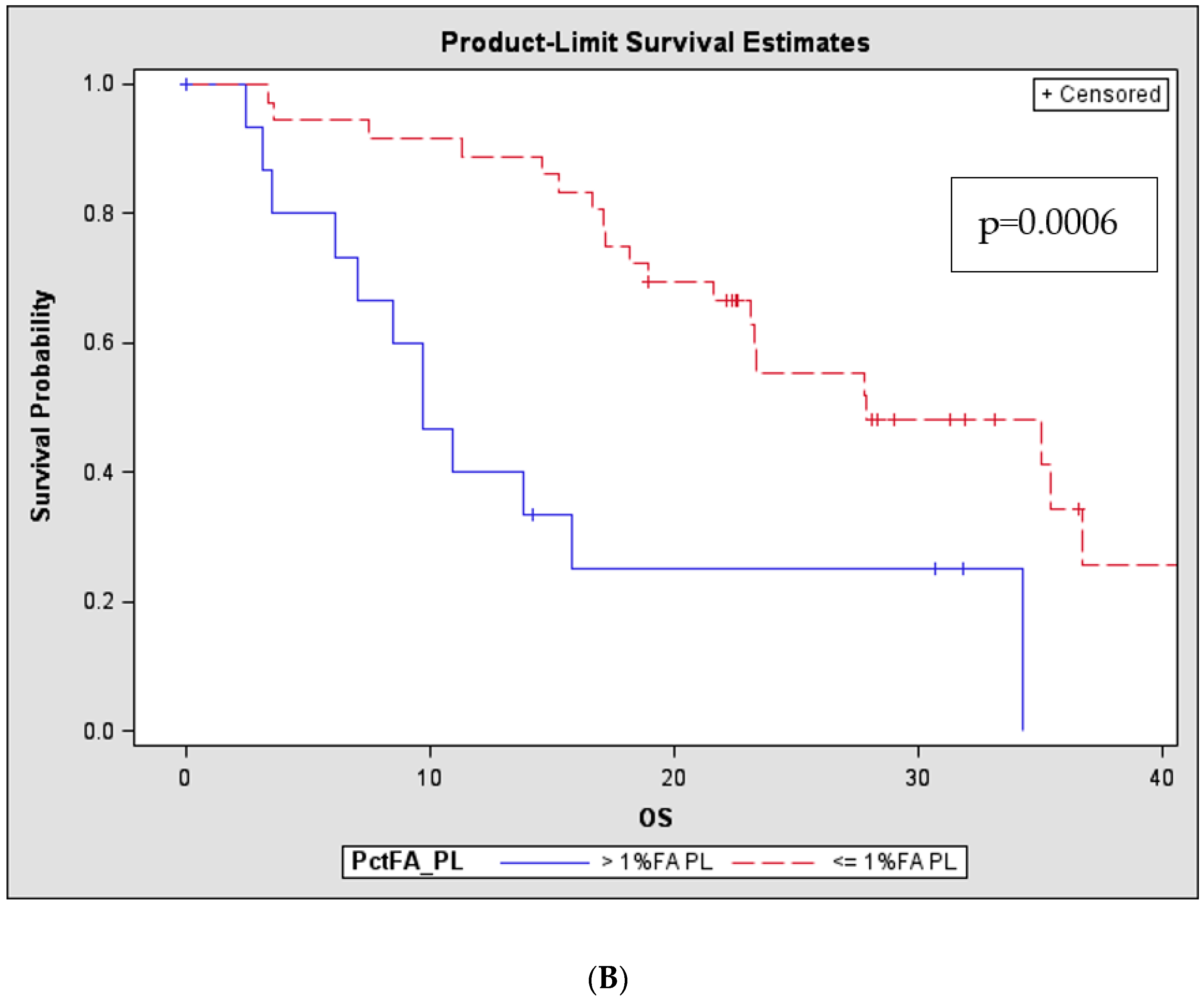
2.2. Patients with Higher Number and Tumour Burden of PL Mutations Have Significantly Shorter Survival
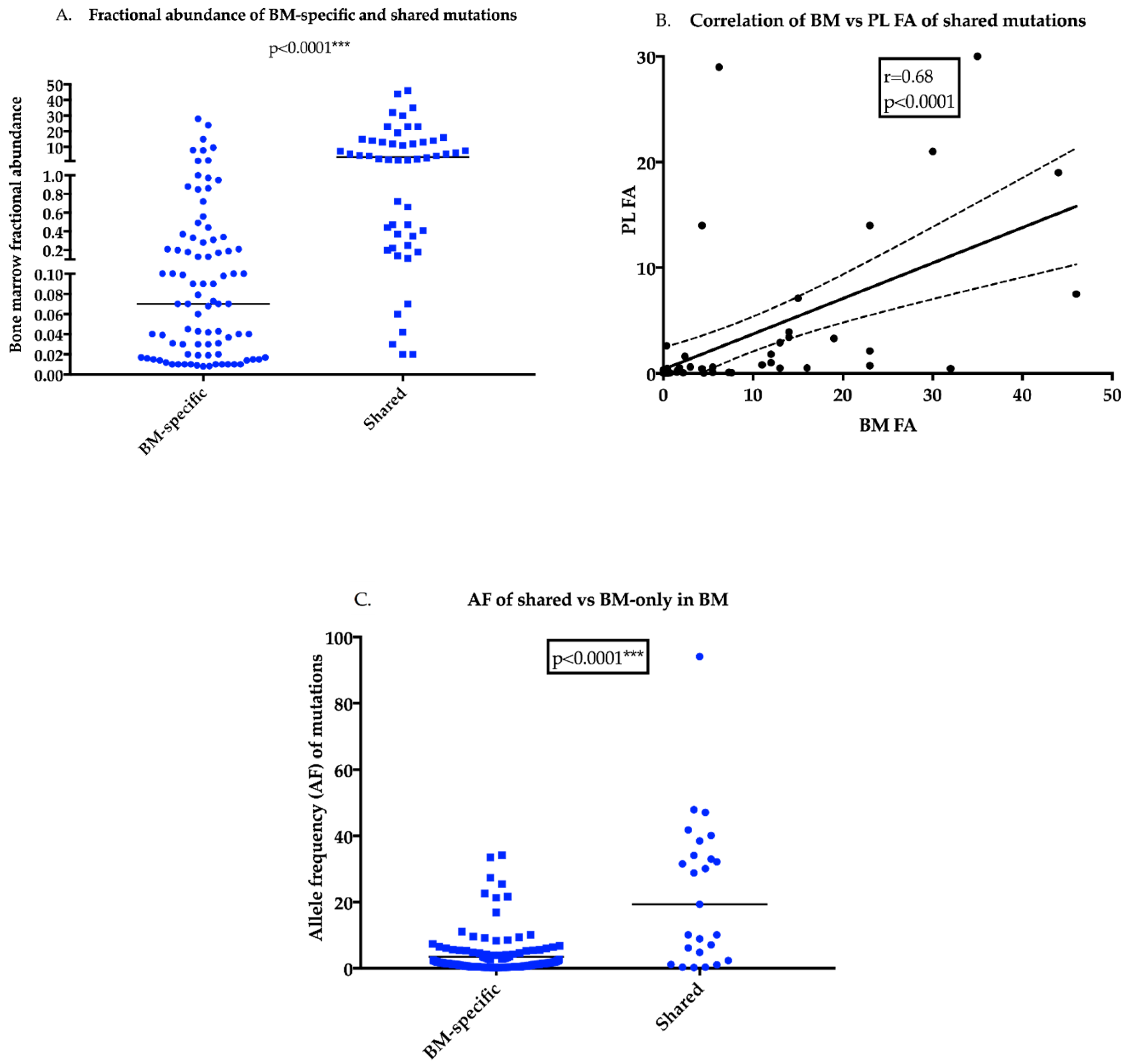
2.3. Clonal Mutations in the BM are More Likely to Be Present in the PL
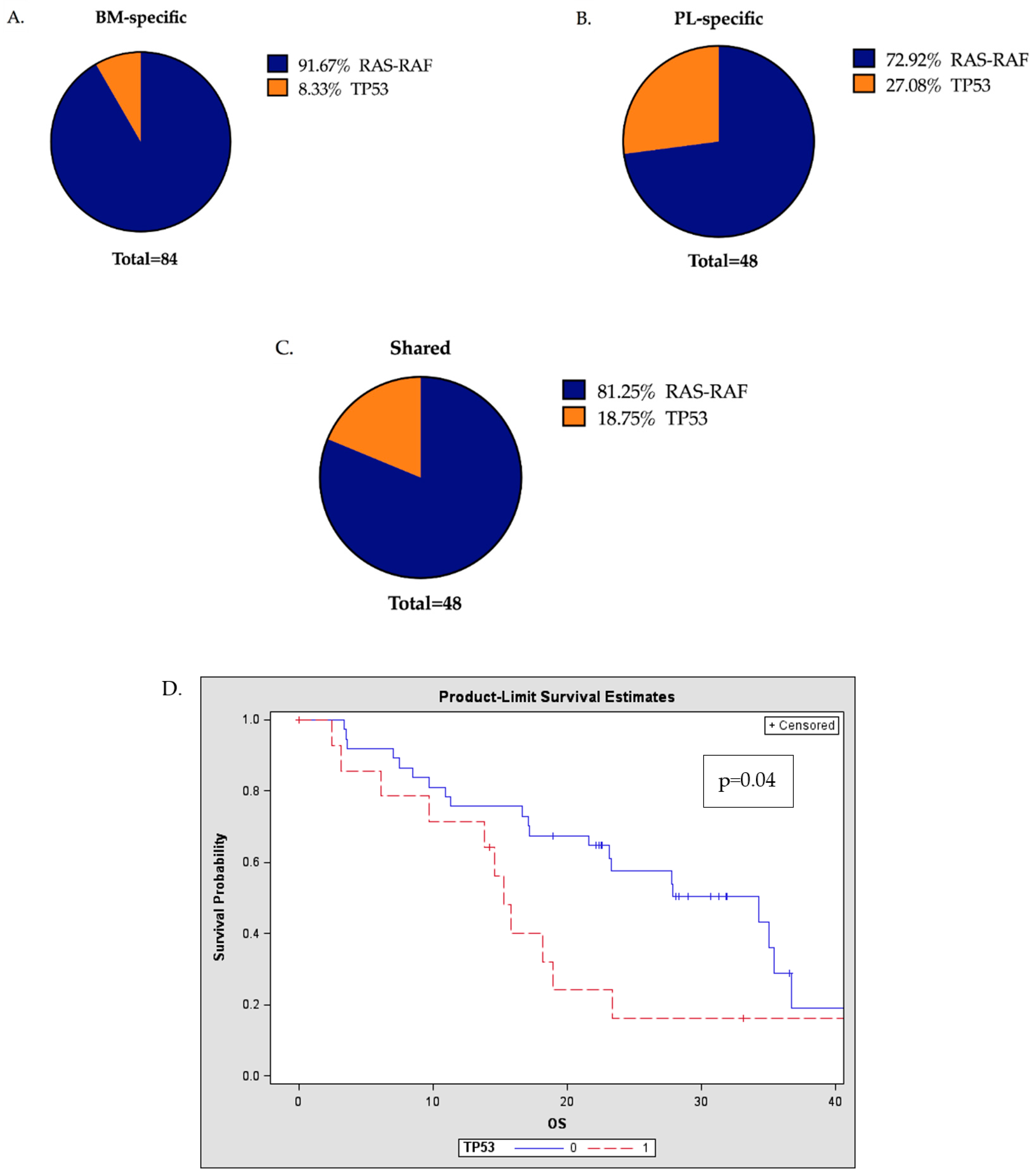
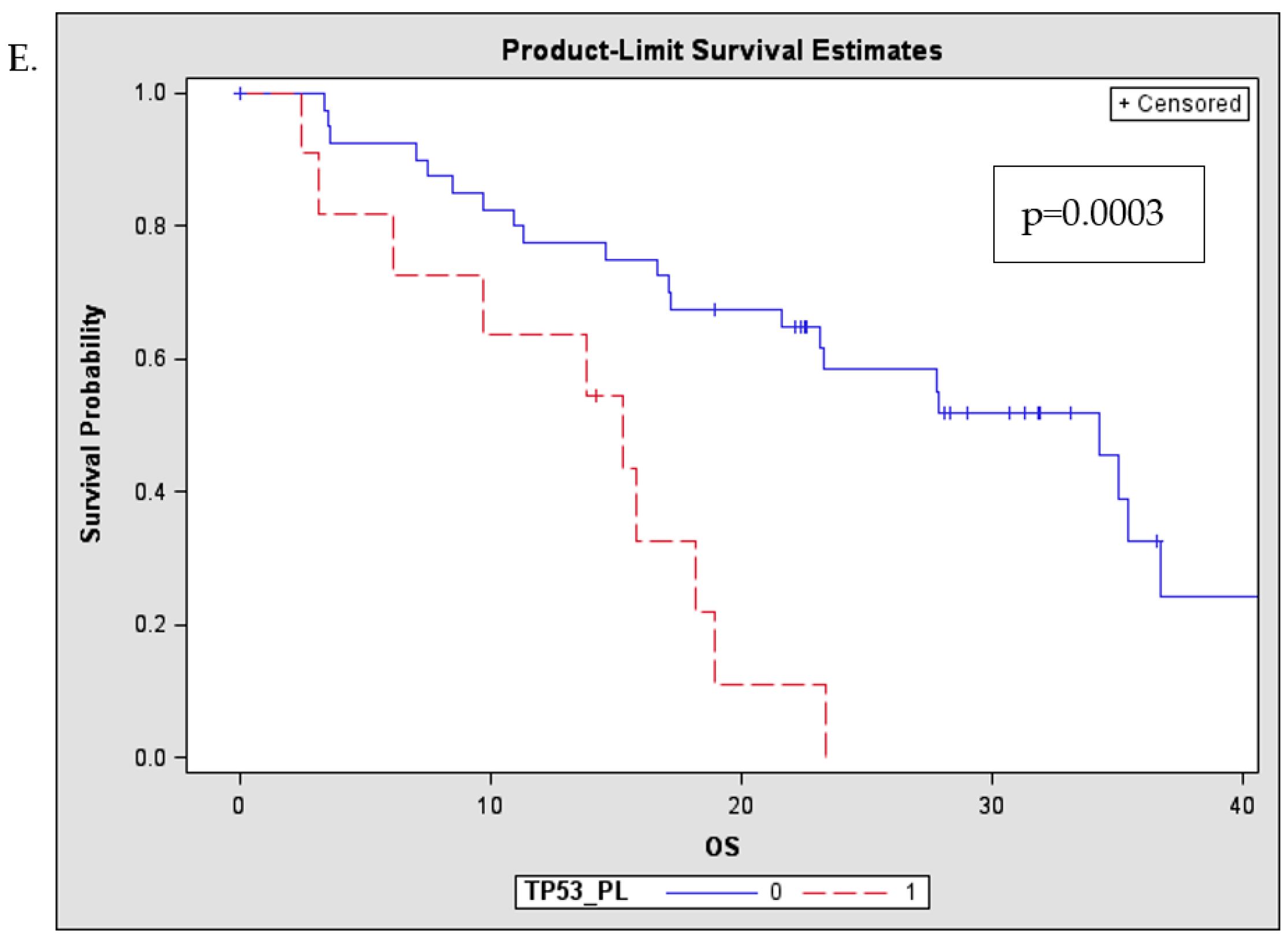
2.4. TP53 Mutations are Frequently Detected in the Plasma of MM Patients
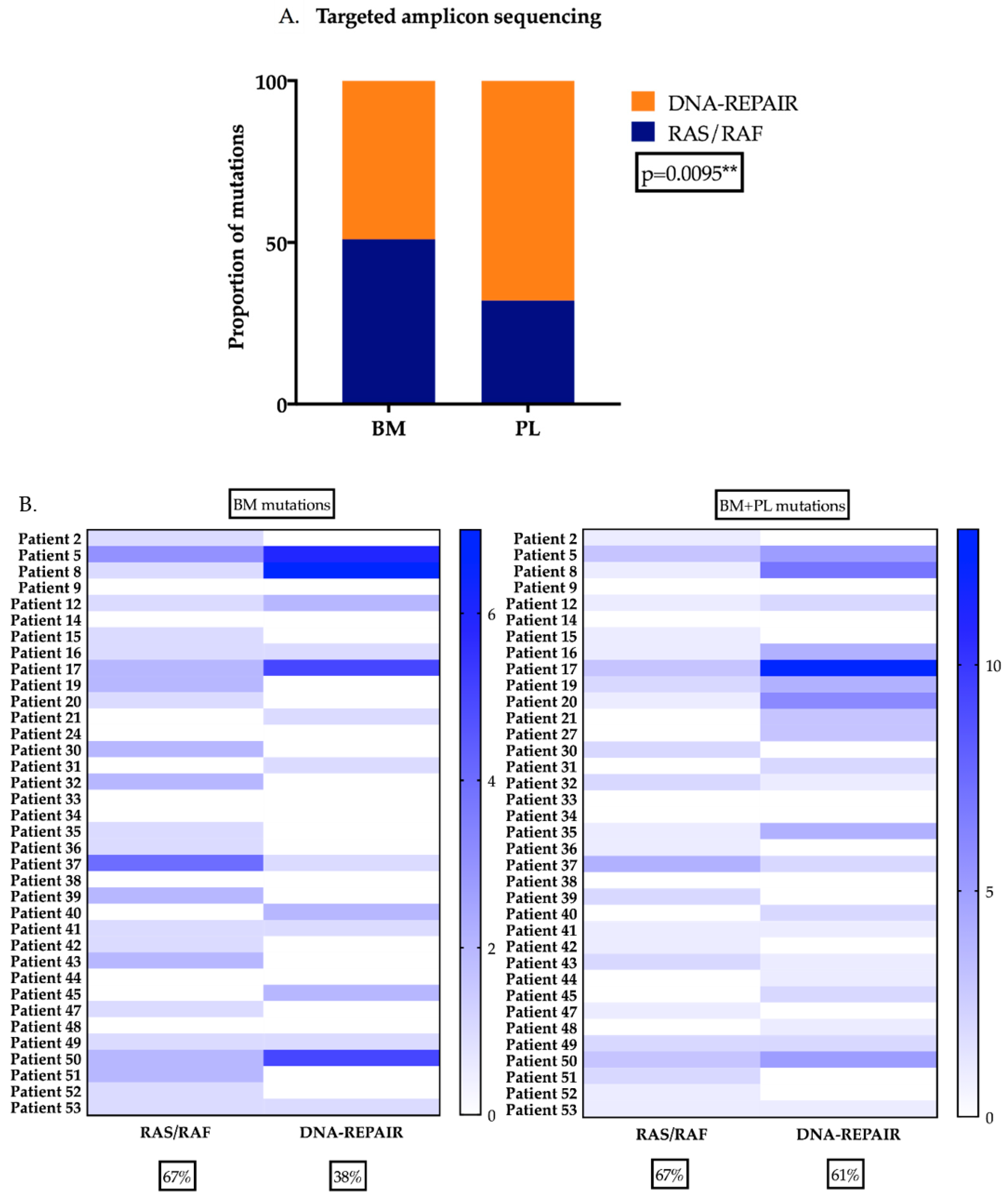
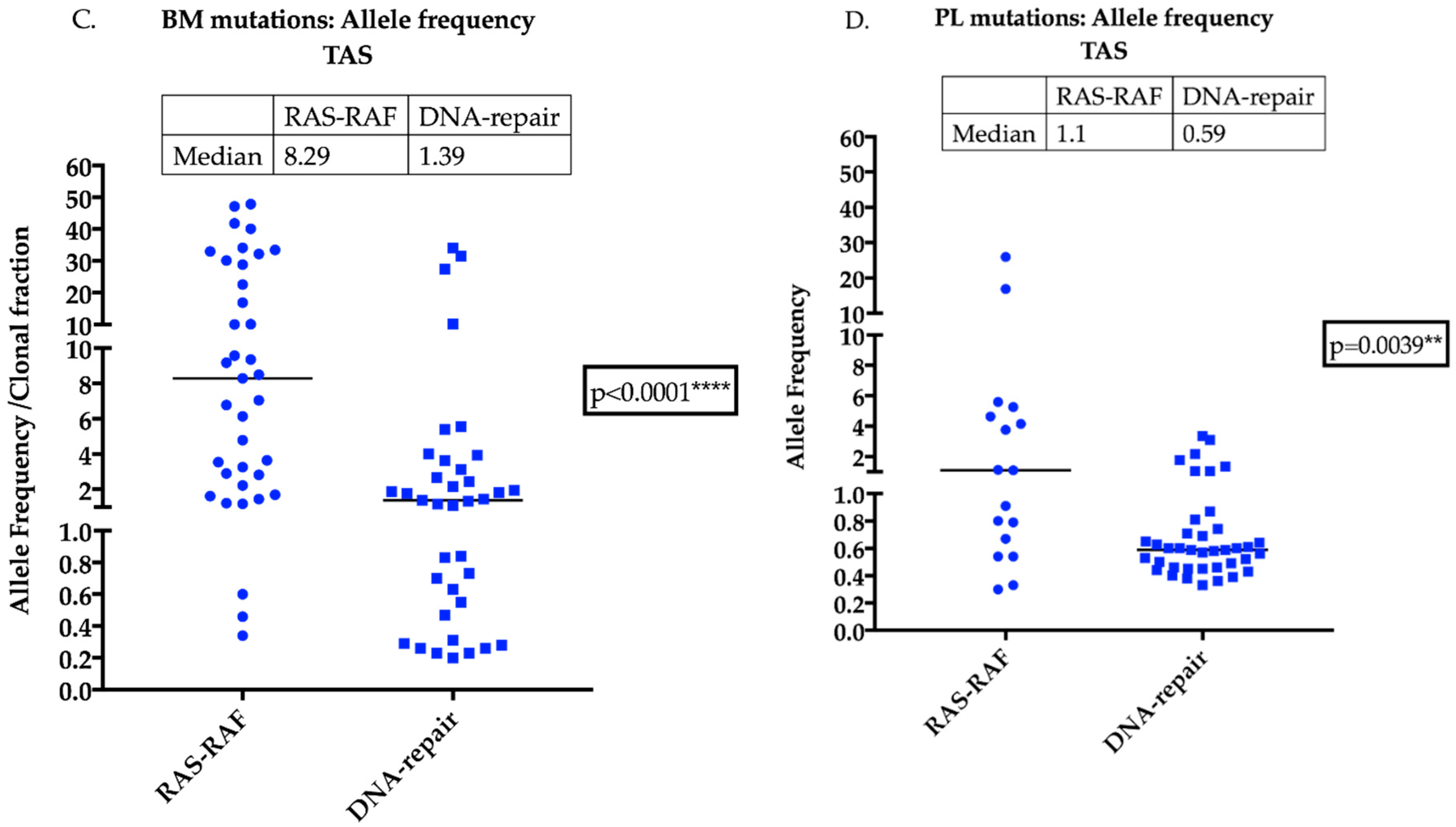
2.5. DNA-Repair Gene Variants are Present at Higher Levels in the Plasma of MM Patients
3. Discussion
4. Materials and Methods
4.1. Patient Population
4.2. Peripheral Blood (PB) Collection and Processing
4.3. Isolation of Mononuclear Cells and MM Cells
4.4. Genomic DNA Extraction From BM Aspirates/ Trephines
4.5. OnTargetTM Mutation Detection (OMD) Platform
4.6. Targeted amplicon sequencing
4.7. Statistical analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| MM | multiple myeloma |
| BM | bone marrow |
| PL | plasma |
| ctDNA | cell free tumour DNA |
| ND | newly diagnosed |
| RR | relapsed and/or refractory |
| OMD | OnTargetTM mutation detection platform |
| TAS | targeted amplicon sequencing |
| FA | fractional abundance |
| PFS | progression-free survival |
| OS | overall survival |
| AF | allele frequency |
References
- Kuehl, W.M.; Bergsagel, P.L. Early genetic events provide the basis for a clinical classification of multiple myeloma. ASH Educ. Program Book 2005, 2005, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients with Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.S.; He, J.; Tytarenko, R.; Deshpande, S.; Patel, P.; Bailey, M.; Stein, C.K.; Stephens, O.; Weinhold, N.; Petty, N.; et al. Bi-allelic inactivation is more prevalent at relapse in multiple myeloma, identifying RB1 as an independent prognostic marker. Blood Cancer J. 2017, 7, e535. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Ryland, G.L.; Jones, K.; Chin, M.; Markham, J.; Aydogan, E.; Kankanige, Y.; Caruso, M.; Guinto, J.; Dickinson, M.; Prince, H.M.; et al. Novel genomic findings in multiple myeloma identified through routine diagnostic sequencing. J. Clin. Pathol. 2018, 71, 895–899. [Google Scholar] [CrossRef]
- Egan, J.B.; Kortuem, K.M.; Kurdoglu, A.; Izatt, T.; Aldrich, J.; Reiman, R.; Phillips, L.; Baker, A.; Shi, C.X.; Schmidt, J.; et al. Extramedullary myeloma whole genome sequencing reveals novel mutations in Cereblon, p.roteasome subunit G2 and the glucocorticoid receptor in multi drug resistant disease. Br. J. Haematol. 2013, 161, 748–751. [Google Scholar] [CrossRef]
- Egan, J.B.; Shi, C.X.; Tembe, W.; Christoforides, A.; Kurdoglu, A.; Sinari, S.; Middha, S.; Asmann, Y.; Schmidt, J.; Braggio, E.; et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, e.volution, a.nd clonal tides. Blood 2012, 120, 1060–1066. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Sirdesai, S.; Chen, M.; Khong, T.; Spencer, A. Circulating Tumour DNA Analysis for Tumour Genome Characterisation and Monitoring Disease Burden in Extramedullary Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 1858. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Jones, J.R.; Lenive, O.; Peterson, E.A.; Brioli, A.; Murison, A.; Wardell, C.P.; Kaiser, M.F.; Proszek, P.; Boyle, E.; et al. Spatiotemporal Analysis of Intraclonal Heterogeneity in Multiple Myeloma: Unravelling the Impact of Treatment and the Propagating Capacity of Subclones Using Whole Exome Sequencing. Blood 2015, 126, e371. [Google Scholar]
- De Haart, S.J.; Willems, S.M.; Mutis, T.; Koudijs, M.J.; van Blokland, M.T.; Lokhorst, H.M.; de Weger, R.A.; Minnema, M.C. Comparison of intramedullary myeloma and corresponding extramedullary soft tissue plasmacytomas using genetic mutational panel analyses. Blood Cancer J. 2016, 6, e426. [Google Scholar] [CrossRef]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia 2017, 31, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Morley, R.; Khong, T.; Kalff, A.; Bergin, K.; Hocking, J.; Savvidou, I.; Bowen, K.M.; Ramachandran, M.; Choi, K.; et al. Monitoring tumour burden and therapeutic response through analysis of circulating tumour DNA and extracellular RNA in multiple myeloma patients. Leukemia 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Kwapisz, D. The first liquid biopsy test approved. Is it a new era of mutation testing for non-small cell lung cancer? Ann. Transl. Med. 2017, 5, e46. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Loehr, A.; Ashby, C.; Tytarenko, R.; Deshpande, S.; Sun, J.; Fedorchak, K.; Mughal, T.; Davies, F.E.; Walker, B.; et al. Loss of heterozygosity as a marker of homologous repair deficiency in multiple myeloma: A role for PARP inhibition? Leukemia 2018, 32, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.B.; Gutierrez, N.C. Targeting Ongoing DNA Damage in Multiple Myeloma: Effects of DNA Damage Response Inhibitors on Plasma Cell Survival. Front. Oncol. 2017, 7, e8. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Alagpulinsa, D.A.; Ayyadevara, S.; Yaccoby, S.; Shmookler Reis, R.J. A Cyclin-Dependent Kinase Inhibitor, D.inaciclib, I.mpairs Homologous Recombination and Sensitizes Multiple Myeloma Cells to PARP Inhibition. Mol. Cancer 2016, 15, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, D.; et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Kidess, E.; Heirich, K.; Wiggin, M.; Vysotskaia, V.; Visser, B.C.; Marziali, A.; Wiedenmann, B.; Norton, J.A.; Lee, M.; Jeffrey, S.; et al. Mutation profiling of tumor DNA from plasma and tumor tissue of colorectal cancer patients with a novel, h.igh-sensitivity multiplexed mutation detection platform. Oncotarget 2015, 6, 2549–2561. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mithraprabhu, S.; Hocking, J.; Ramachandran, M.; Choi, K.; Klarica, D.; Khong, T.; Reynolds, J.; Spencer, A. DNA-Repair Gene Mutations Are Highly Prevalent in Circulating Tumour DNA from Multiple Myeloma Patients. Cancers 2019, 11, 917. https://doi.org/10.3390/cancers11070917
Mithraprabhu S, Hocking J, Ramachandran M, Choi K, Klarica D, Khong T, Reynolds J, Spencer A. DNA-Repair Gene Mutations Are Highly Prevalent in Circulating Tumour DNA from Multiple Myeloma Patients. Cancers. 2019; 11(7):917. https://doi.org/10.3390/cancers11070917
Chicago/Turabian StyleMithraprabhu, Sridurga, Jay Hocking, Malarmathy Ramachandran, Kawa Choi, Daniela Klarica, Tiffany Khong, John Reynolds, and Andrew Spencer. 2019. "DNA-Repair Gene Mutations Are Highly Prevalent in Circulating Tumour DNA from Multiple Myeloma Patients" Cancers 11, no. 7: 917. https://doi.org/10.3390/cancers11070917
APA StyleMithraprabhu, S., Hocking, J., Ramachandran, M., Choi, K., Klarica, D., Khong, T., Reynolds, J., & Spencer, A. (2019). DNA-Repair Gene Mutations Are Highly Prevalent in Circulating Tumour DNA from Multiple Myeloma Patients. Cancers, 11(7), 917. https://doi.org/10.3390/cancers11070917