AP-1 Transcription Factors as Regulators of Immune Responses in Cancer



Abstract
1. Introduction
2. Activator Protein-1 (AP-1) Transcription Factors
2.1. Regulation of Immune Response by AP-1 in Genetically Modified Mice
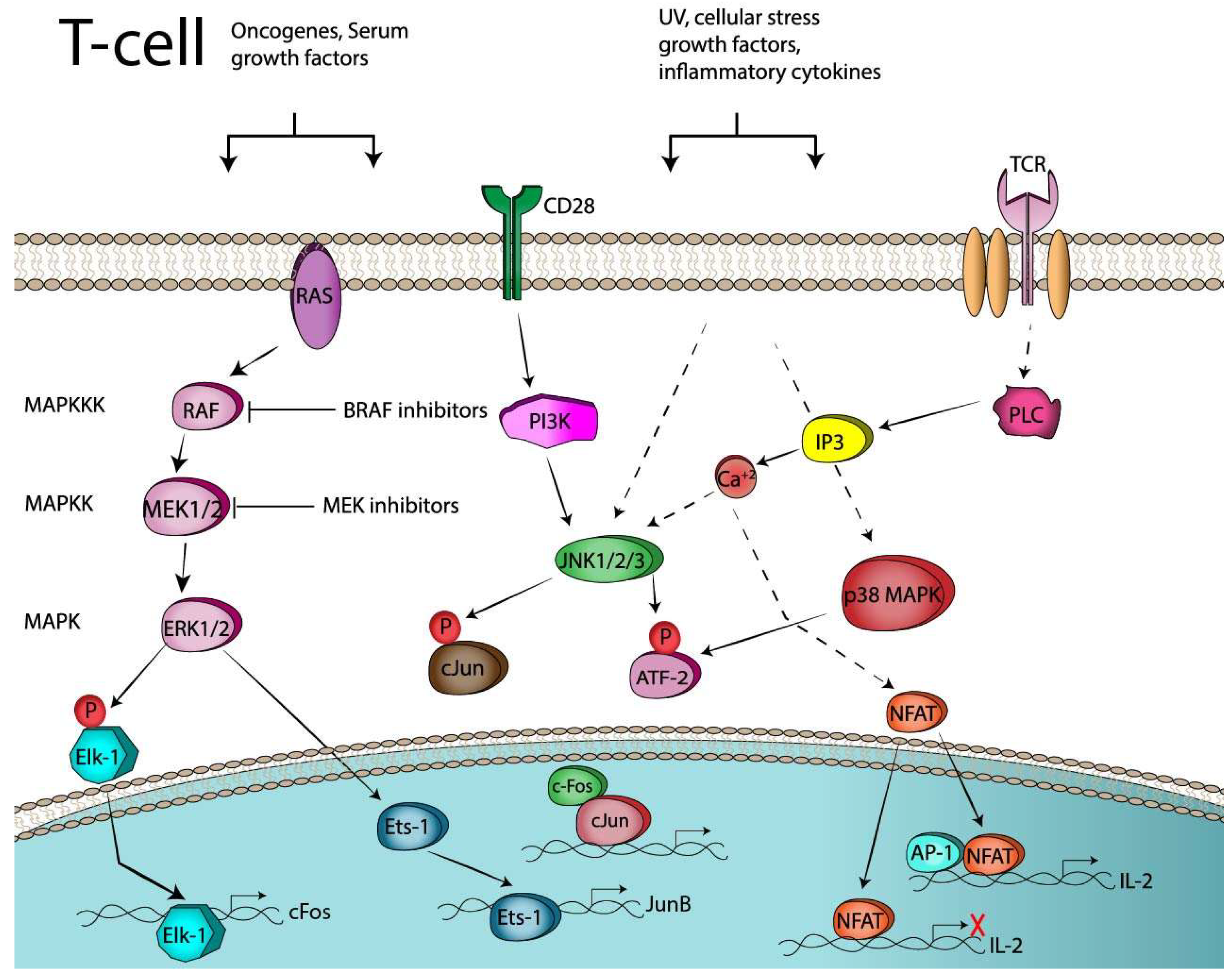
2.2. AP-1 and T-Cell Activation
2.3. AP-1 and T-Cell Anergy or Exhaustion
3. AP-1 and Immune Checkpoint Regulation
3.1. Co-Stimulatory Molecules and AP-1 Transcription Factors
3.1.1. CD28
3.1.2. CD40/CD40L
3.1.3. ICOS/4-1BB
3.2. Co-Inhibitory Molecules
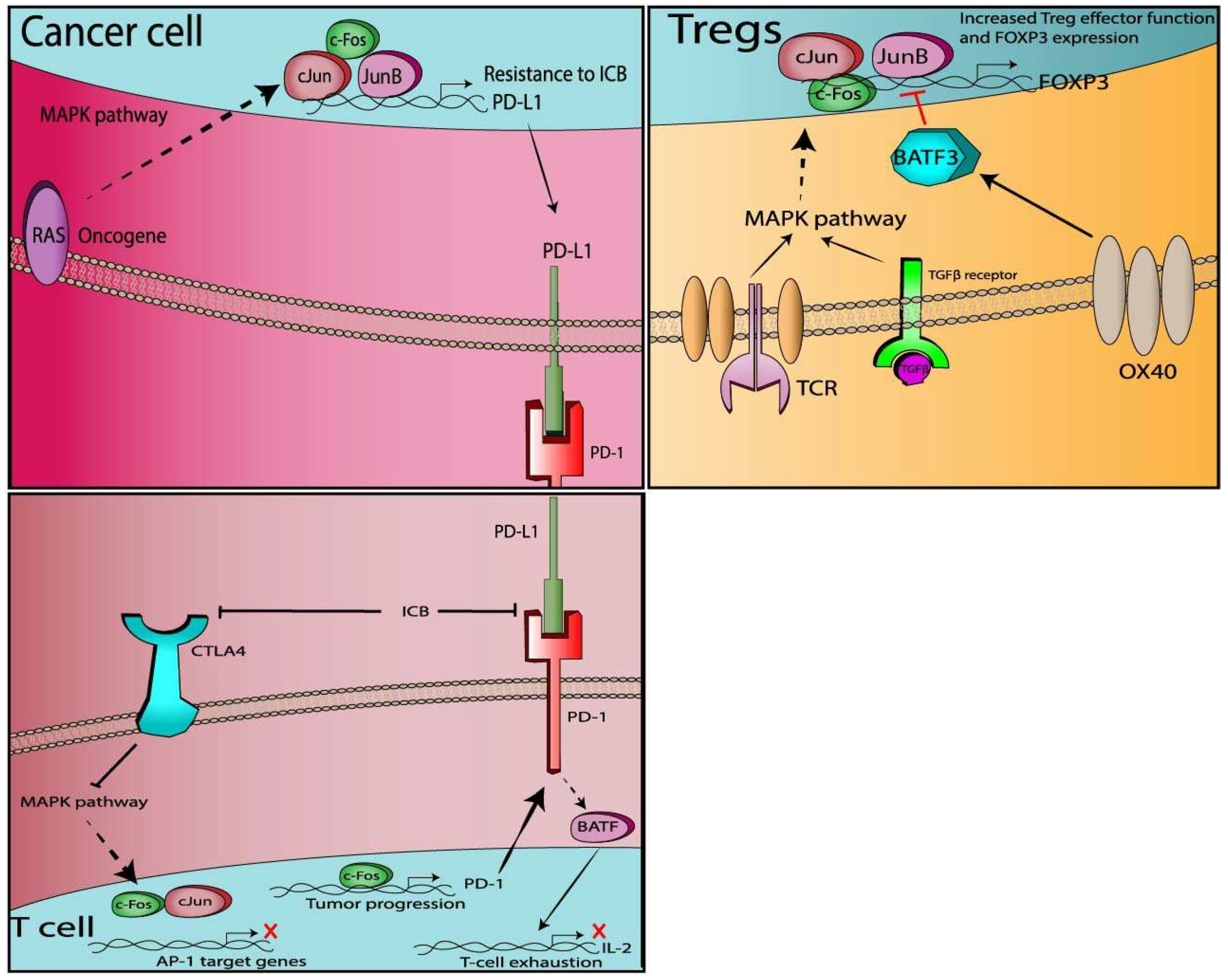
3.2.1. PD-1
3.2.2. PD-L1
3.2.3. CTLA-4
3.2.4. Tregs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Roy, A.; Masson, E.; Chen, T.T.; Humphrey, R.; Weber, J.S. Exposure-response relationships of the efficacy and safety of ipilimumab in patients with advanced melanoma. Clin. Cancer Res. 2013, 19, 3977–3986. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vetizou, M.; Daillere, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and-extrinsic factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Dunn, I.S.; Durda, P.J.; Butera, D.; Rose, L.B.; Haggerty, T.J.; Benson, E.M.; Kurnick, J.T. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol. Cancer Res. 2006, 4, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [PubMed]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Mimura, K.; Shiraishi, K.; Mueller, A.; Izawa, S.; Kua, L.F.; So, J.; Yong, W.P.; Fujii, H.; Seliger, B.; Kiessling, R.; et al. The MAPK pathway is a predominant regulator of HLA-A expression in esophageal and gastric cancer. J. Immunol. 2013, 191, 6261–6272. [Google Scholar] [CrossRef]
- Brea, E.J.; Oh, C.Y.; Manchado, E.; Budhu, S.; Gejman, R.S.; Mo, G.; Mondello, P.; Han, J.E.; Jarvis, C.A.; Ulmert, D.; et al. Kinase regulation of human MHC class I molecule expression on cancer cells. Cancer Immunol. Res. 2016, 4, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP kinase inhibition promotes T Cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef]
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: A review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019, 11, 1758835919830826. [Google Scholar] [CrossRef]
- Tawbi, H.A.-H.; Amaria, R.N.; Glitza, I.C.; Milton, D.; Hwu, W.-J.; Patel, S.P.; Wong, M.K.K.; Yee, C.; Woodman, S.E.; McQuade, J.L.; et al. Safety and preliminary activity data from a single center phase II study of triplet combination of nivolumab (N) with dabrafenib (D) and trametinib (T) [trident] in patients (Pts) with BRAF-mutated metastatic melanoma (MM). J. Clin. Oncol. 2018, 36, 9560. [Google Scholar] [CrossRef]
- Dummer, R.; Fernández, A.M.A.; Hansson, J.; Larkin, J.M.G.; Long, G.V.; Gasal, E.; Kaper, M.; Upalawanna, A.; Mookerjee, B.; Atkinson, V. Preliminary findings from part 1 of COMBI-i: A phase III study of anti–PD-1 antibody PDR001 combined with dabrafenib (D) and trametinib (T) in previously untreated patients (pts) with advanced BRAF V600-mutant melanoma. J. Clin. Oncol. 2018, 36, 189. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Foletta, V.C.; Segal, D.H.; Cohen, D.R. Transcriptional regulation in the immune system: All roads lead to AP-1. J. Leukoc. Biol. 1998, 63, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Eferl, R. Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 2005, 208, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Haslinger, A.; Karin, M.; Tjian, R. Activation of transcription by two factors that bind promoter and enhancer sequences of the human metallothionein gene and SV40. Nature 1987, 325, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Angel, P.; Imagawa, M.; Chiu, R.; Stein, B.; Imbra, R.J.; Rahmsdorf, H.J.; Jonat, C.; Herrlich, P.; Karin, M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell 1987, 49, 729–739. [Google Scholar] [CrossRef]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y.; Nathans, D. The basic region of Fos mediates specific DNA binding. EMBO J. 1989, 8, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Zerial, M.; Toschi, L.; Ryseck, R.P.; Schuermann, M.; Muller, R.; Bravo, R. The product of a novel growth factor activated gene, fos B, interacts with JUN proteins enhancing their DNA binding activity. EMBO J. 1989, 8, 805–813. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Georgopoulos, K.; Greenberg, M.E.; Leder, P. C-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell 1988, 55, 917–924. [Google Scholar] [CrossRef]
- Chiu, R.; Angel, P.; Karin, M. Jun-B differs in its biological properties from, and is a negative regulator of, c-Jun. Cell 1989, 59, 979–986. [Google Scholar] [CrossRef]
- Schutte, J.; Viallet, J.; Nau, M.; Segal, S.; Fedorko, J.; Minna, J. Jun-B inhibits and c-fos stimulates the transforming and trans-activating activities of c-jun. Cell 1989, 59, 987–997. [Google Scholar] [CrossRef]
- Li, B.; Tournier, C.; Davis, R.J.; Flavell, R.A. Regulation of IL-4 expression by the transcription factor Jun-B during T helper cell differentiation. EMBO J. 1999, 18, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Behrens, A.; Sabapathy, K.; Graef, I.; Cleary, M.; Crabtree, G.R.; Wagner, E.F. Jun N-terminal kinase 2 modulates thymocyte apoptosis and T cell activation through c-Jun and nuclear factor of activated T cell (NF-AT). Proc. Natl. Acad. Sci. USA 2001, 98, 1769–1774. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, M.L.; Jacobs, H.; Acton, D.; Verma, I.; Berns, A. Overexpression of the Fos B2 gene in thymocytes causes aberrant development of T cells and thymic epithelial cells. Oncogene 1997, 14, 1083–1091. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huse, M. The T-cell-receptor signaling network. J. Cell Sci. 2009, 122, 1269–1273. [Google Scholar] [CrossRef]
- Linsley, P.S.; Ledbetter, J.A. The role of the CD28 receptor during T cell responses to antigen. Annu. Rev. Immunol. 1993, 11, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Marais, R.; Wynne, J.; Treisman, R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 1993, 73, 381–393. [Google Scholar] [CrossRef]
- Hibi, M.; Lin, A.; Smeal, T.; Minden, A.; Karin, M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993, 7, 2135–2148. [Google Scholar] [CrossRef]
- Spangler, B.; Vardimon, L.; Bosserhoff, A.K.; Kuphal, S. Post-transcriptional regulation controlled by E-cadherin is important for c-Jun activity in melanoma. Pigment. Cell Melanoma Res. 2011, 24, 148–164. [Google Scholar] [CrossRef] [PubMed]
- Edmead, C.E.; Patel, Y.I.; Wilson, A.; Boulougouris, G.; Hall, N.D.; Ward, S.G.; Sansom, D.M. Induction of activator protein (AP)-1 and nuclear factor-kappaB by CD28 stimulation involves both phosphatidylinositol 3-kinase and acidic sphingomyelinase signals. J. Immunol. 1996, 157, 3290–3297. [Google Scholar] [PubMed]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472. [Google Scholar] [CrossRef] [PubMed]
- Macian, F.; Lopez-Rodriguez, C.; Rao, A. Partners in transcription: NFAT and AP-1. Oncogene 2001, 20, 2476–2489. [Google Scholar] [CrossRef] [PubMed]
- McGuire, K.L.; Iacobelli, M. Involvement of Rel, Fos, and Jun proteins in binding activity to the IL-2 promoter CD28 response element/AP-1 sequence in human T cells. J. Immunol. 1997, 159, 1319–1327. [Google Scholar] [PubMed]
- Schwartz, R.H. T cell anergy. Annu. Rev. Immunol. 2003, 21, 305–334. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Martinez, G.J.; Pereira, R.M.; Aijo, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef]
- Macian, F.; Garcia-Cozar, F.; Im, S.H.; Horton, H.F.; Byrne, M.C.; Rao, A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 2002, 109, 719–731. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Fields, P.E.; Gajewski, T.F.; Fitch, F.W. Blocked Ras activation in anergic CD4+ T cells. Science 1996, 271, 1276–1278. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Whaley, C.D.; Mondino, A.; Mueller, D.L. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science 1996, 271, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Mognol, G.P.; González-Avalos, E.; Ghosh, S.; Spreafico, R.; Gudlur, A.; Rao, A.; Damoiseaux, R.; Hogan, P.G. Targeting the NFAT: AP-1 transcriptional complex on DNA with a small-molecule inhibitor. Proc. Natl. Acad. Sci. USA 2019, 116, 9959–9968. [Google Scholar] [PubMed]
- Rizvi, N.A.; Mazieres, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol. 2015, 16, 257–265. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.A.; St John, T.; Allison, J.P. The murine homologue of the T lymphocyte antigen CD28. Molecular cloning and cell surface expression. J. Immunol. 1990, 144, 3201–3210. [Google Scholar] [PubMed]
- Shahinian, A.; Pfeffer, K.; Lee, K.P.; Kundig, T.M.; Kishihara, K.; Wakeham, A.; Kawai, K.; Ohashi, P.S.; Thompson, C.B.; Mak, T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science 1993, 261, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Granelli-Piperno, A.; Nolan, P. Nuclear transcription factors that bind to elements of the IL-2 promoter. Induction requirements in primary human T cells. J. Immunol. 1991, 147, 2734–2739. [Google Scholar] [PubMed]
- Rincon, M.; Flavell, R.A. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J. 1994, 13, 4370–4381. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Jacinto, E.; Hibi, M.; Kallunki, T.; Karin, M.; Ben-Neriah, Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell 1994, 77, 727–736. [Google Scholar] [CrossRef]
- Chatta, G.S.; Spies, A.G.; Chang, S.; Mize, G.J.; Linsley, P.S.; Ledbetter, J.A.; Morris, D.R. Differential regulation of proto-oncogenes c-jun and c-fos in T lymphocytes activated through CD28. J. Immunol. 1994, 153, 5393–5401. [Google Scholar] [PubMed]
- Janardhan, S.V.; Praveen, K.; Marks, R.; Gajewski, T.F. Evidence implicating the Ras pathway in multiple CD28 costimulatory functions in CD4+ T cells. PLoS ONE 2011, 6, e24931. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Whaley, C.D.; Bonnevier, J.L.; Mondino, A.; Martin, M.E.; Aagaard-Tillery, K.M.; Mueller, D.L. CD28 signaling augments Elk-1-dependent transcription at the c-fos gene during antigen stimulation. J. Immunol. 2001, 167, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Van Kooten, C.; Banchereau, J. Functions of CD40 on B cells, dendritic cells and other cells. Curr. Opin. Immunol. 1997, 9, 330–337. [Google Scholar] [CrossRef]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef]
- Bishop, G.A.; Moore, C.R.; Xie, P.; Stunz, L.L.; Kraus, Z.J. TRAF proteins in CD40 signaling. Adv. Exp. Med. Biol. 2007, 597, 131–151. [Google Scholar] [CrossRef]
- Georgopoulos, N.T.; Steele, L.P.; Thomson, M.J.; Selby, P.J.; Southgate, J.; Trejdosiewicz, L.K. A novel mechanism of CD40-induced apoptosis of carcinoma cells involving TRAF3 and JNK/AP-1 activation. Cell Death Differ. 2006, 13, 1789–1801. [Google Scholar] [CrossRef]
- Tsytsykova, A.V.; Tsitsikov, E.N.; Geha, R.S. The CD40L promoter contains nuclear factor of activated T cells-binding motifs which require AP-1 binding for activation of transcription. J. Biol. Chem. 1996, 271, 3763–3770. [Google Scholar] [CrossRef]
- Afford, S.C.; Ahmed-Choudhury, J.; Randhawa, S.; Russell, C.; Youster, J.; Crosby, H.A.; Eliopoulos, A.; Hubscher, S.G.; Young, L.S.; Adams, D.H. CD40 activation-induced, Fas-dependent apoptosis and NF-kappaB/AP-1 signaling in human intrahepatic biliary epithelial cells. FASEB J. 2001, 15, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Baccam, M.; Woo, S.Y.; Vinson, C.; Bishop, G.A. CD40-mediated transcriptional regulation of the IL-6 gene in B lymphocytes: Involvement of NF-kappa B, AP-1, and C/EBP. J. Immunol. 2003, 170, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Vanden Bush, T.J.; Bishop, G.A. TLR7 and CD40 cooperate in IL-6 production via enhanced JNK and AP-1 activation. Eur. J. Immunol. 2008, 38, 400–409. [Google Scholar] [CrossRef] [PubMed]
- D’Aversa, T.G.; Eugenin, E.A.; Berman, J.W. CD40-CD40 ligand interactions in human microglia induce CXCL8 (interleukin-8) secretion by a mechanism dependent on activation of ERK1/2 and nuclear translocation of nuclear factor-kappaB (NFkappaB) and activator protein-1 (AP-1). J. Neurosci. Res. 2008, 86, 630–639. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Coyle, A.J.; Lehar, S.; Lloyd, C.; Tian, J.; Delaney, T.; Manning, S.; Nguyen, T.; Burwell, T.; Schneider, H.; Gonzalo, J.A.; et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity 2000, 13, 95–105. [Google Scholar] [CrossRef]
- Watanabe, M.; Nakajima, S.; Ohnuki, K.; Ogawa, S.; Yamashita, M.; Nakayama, T.; Murakami, Y.; Tanabe, K.; Abe, R. AP-1 is involved in ICOS gene expression downstream of TCR/CD28 and cytokine receptor signaling. Eur. J. Immunol. 2012, 42, 1850–1862. [Google Scholar] [CrossRef]
- Kim, J.O.; Kim, H.W.; Baek, K.M.; Kang, C.Y. NF-kappaB and AP-1 regulate activation-dependent CD137 (4-1BB) expression in T cells. FEBS Lett. 2003, 541, 163–170. [Google Scholar] [CrossRef]
- Kwon, B.S.; Kozak, C.A.; Kim, K.K.; Pickard, R.T. Genomic organization and chromosomal localization of the T-cell antigen 4-1BB. J. Immunol. 1994, 152, 2256–2262. [Google Scholar]
- Kim, J.D.; Kim, C.H.; Kwon, B.S. Regulation of mouse 4-1BB expression: Multiple promoter usages and a splice variant. Mol. Cells 2011, 31, 141–149. [Google Scholar] [CrossRef]
- Arasanz, H.; Gato-Cañas, M.; Zuazo, M.; Ibañez-Vea, M.; Breckpot, K.; Kochan, G.; Escors, D. PD1 signal transduction pathways in T cells. Oncotarget 2017, 8, 51936–51945. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed]
- Quigley, M.; Pereyra, F.; Nilsson, B.; Porichis, F.; Fonseca, C.; Eichbaum, Q.; Julg, B.; Jesneck, J.L.; Brosnahan, K.; Imam, S.; et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010, 16, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Deng, A.; Liu, H.; Ge, G.; Liu, X. Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15419–15424. [Google Scholar] [CrossRef]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Zerdes, I.; Matikas, A.; Bergh, J.; Rassidakis, G.Z.; Foukakis, T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: Biology and clinical correlations. Oncogene 2018, 37, 4639–4661. [Google Scholar] [CrossRef]
- Mathas, S.; Hinz, M.; Anagnostopoulos, I.; Krappmann, D.; Lietz, A.; Jundt, F.; Bommert, K.; Mechta-Grigoriou, F.; Stein, H.; Dorken, B.; et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002, 21, 4104–4113. [Google Scholar] [CrossRef]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef]
- Fang, W.; Zhang, J.; Hong, S.; Zhan, J.; Chen, N.; Qin, T.; Tang, Y.; Zhang, Y.; Kang, S.; Zhou, T.; et al. EBV-driven LMP1 and IFN-gamma up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget 2014, 5, 12189–12202. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhou, J.; Giobbie-Hurder, A.; Wargo, J.; Hodi, F.S. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin. Cancer Res. 2013, 19, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Takano, A.; Teramoto, K.; Daigo, Y. RAS-mitogen-activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PLoS ONE 2016, 11, e0166626. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The interaction properties of costimulatory molecules revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25(+) CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef]
- Calvo, C.R.; Amsen, D.; Kruisbeek, A.M. Cytotoxic T lymphocyte antigen 4 (CTLA-4) interferes with extracellular signal-regulated kinase (ERK) and Jun NH2-terminal kinase (JNK) activation, but does not affect phosphorylation of T cell receptor zeta and ZAP70. J. Exp. Med. 1997, 186, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Olsson, C.; Riesbeck, K.; Dohlsten, M.; Michaelsson, E. CTLA-4 ligation suppresses CD28-induced NF-kappaB and AP-1 activity in mouse T cell blasts. J. Biol. Chem. 1999, 274, 14400–14405. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.H.; Rincon, M.; McCoy, K.D.; Le Gros, G. CTLA4 ligation attenuates AP-1, NFAT and NF-kappaB activity in activated T cells. Eur. J. Immunol. 1999, 29, 838–844. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2016, 27, 109. [Google Scholar] [CrossRef]
- Mantel, P.Y.; Ouaked, N.; Ruckert, B.; Karagiannidis, C.; Welz, R.; Blaser, K.; Schmidt-Weber, C.B. Molecular mechanisms underlying FOXP3 induction in human T cells. J. Immunol. 2006, 176, 3593–3602. [Google Scholar] [CrossRef]
- Ogawa, C.; Tone, Y.; Tsuda, M.; Peter, C.; Waldmann, H.; Tone, M. TGF-beta-mediated Foxp3 gene expression is cooperatively regulated by Stat5, Creb, and AP-1 through CNS2. J. Immunol. 2014, 192, 475–483. [Google Scholar] [CrossRef]
- Lee, S.M.; Gao, B.; Fang, D. FoxP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA-binding activity of AP-1. Blood 2008, 111, 3599–3606. [Google Scholar] [CrossRef]
- Bao, R.; Hou, J.; Li, Y.; Bian, J.; Deng, X.; Zhu, X.; Yang, T. Adenosine promotes Foxp3 expression in Treg cells in sepsis model by activating JNK/AP-1 pathway. Am. J. Transl. Res. 2016, 8, 2284–2292. [Google Scholar] [PubMed]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kitani, A.; Stuelten, C.; McGrady, G.; Fuss, I.; Strober, W. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity 2010, 33, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Kim, H.S.; Hwang, S.S.; Lee, G.R. The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Exp. Mol. Med. 2017, 49, e393. [Google Scholar] [CrossRef] [PubMed]
- Hayatsu, N.; Miyao, T.; Tachibana, M.; Murakami, R.; Kimura, A.; Kato, T.; Kawakami, E.; Endo, T.A.; Setoguchi, R.; Watarai, H.; et al. Analyses of a mutant Foxp3 allele reveal BATF as a critical transcription factor in the differentiation and accumulation of tissue regulatory T cells. Immunity 2017, 47, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xiao, X.; Lan, P.; Li, J.; Dou, Y.; Chen, W.; Ishii, N.; Chen, S.; Xia, B.; Chen, K.; et al. OX40 costimulation inhibits Foxp3 expression and Treg induction via BATF3-dependent and independent mechanisms. Cell Rep. 2018, 24, 607–618. [Google Scholar] [CrossRef]
- Koizumi, S.I.; Sasaki, D.; Hsieh, T.H.; Taira, N.; Arakaki, N.; Yamasaki, S.; Wang, K.; Sarkar, S.; Shirahata, H.; Miyagi, M.; et al. JunB regulates homeostasis and suppressive functions of effector regulatory T cells. Nat. Commun. 2018, 9, 5344. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| National Clinical Trials Identifier | Phase | Targeted Therapy/Immunotherapy | Institution | Scheduling | Indications |
|---|---|---|---|---|---|
| NCT02224781 | III | D + T, Ipi + Nivo or Ipi + Nivo, D + T | University of Alabama at Birmingham Cancer Center | Sequential | Melanoma |
| NCT02910700 | II | Nivo + T, with or w/o D | M.D Anderson Cancer Center | Concurrent | Melanoma |
| NCT01940809 | I | Ipi w and w/o D, T and/or Nivo | Brigham and Women’s Hospital | Sequential | Melanoma |
| NCT02130466 | I/II | Pembro + T + D | Several locations, USA | Combination | Melanoma |
| NCT02858921 | II | D + T + Pembro | Several location, Australia | Sequential | Melanoma |
| NCT02625337 | II | Pembro + D + T | Antoni van Leeuwenhoek ziekenhuis Amsterdam, | Concurrent | Melanoma |
| NCT03149029 | II | Pembro + D + T | Beth Israel Deaconess Medical Center | Concurrent | Melanoma |
| NCT02060188 | II | Nivo + Ipi + C | Several locations, USA | Concurrent | Colon Cancer |
| NCT02818023 | I | Pembro + V + C | UPMC Cancer Center Hillman Cancer Center | Concurrent | Melanoma |
| NCT03273153 | III | Atezo + C | University of South Alabama; Mitchell Cancer Institute | Concurrent | Melanoma |
| NCT03013491 | I/II | CX-072 (anti-PD-L1) + V | Several locations, USA | Concurrent | Solid tumors and Lymphoma |
| NCT02968303 | II | V + C, Ipi + Nivo | Several locations, The Netherlands | Sequential | Melanoma |
| Gene Name | Direction of the Interaction | AP-1 Member | Mechanism | Activity | Cell Type | References |
|---|---|---|---|---|---|---|
| CD28 | → (upregulates) | cJun | PI3K, JNK and ERK dependent | T-cell activation | [42,60,61,62,63] | |
| ICOS | ← (upregulates) | Fra-2 and other AP-1 | TCR/CD28 stimulation/ ICOS promoter binding | Expansion of several T helper subsets and Tregs | T-cells | [77] |
| 4-1BB | ← (upregulates) | AP-1 | 4-1BB promoter binding. MEK and JNK dependent. | Co-stimulation of T-cells responses | T-cells | [79,80] |
| PD-1 | → (upregulates) | BATF | Inhibits T-cell function | T-cells in chronic viral infections | [85] | |
| PD-1 | ← (upregulates) | c-Fos | PD-1 promoter binding | Increased tumor burden when expressed in infiltrating T-cells | T-cells in mouse model of lung carcinoma | [86] |
| PD-L1 | ← (upregulates) | cJun, JunB | PD-L1 promoter binding | EBV(+) tumor cells | [91] | |
| PD-L1 | ← (upregulates) | cJun | MAPK dependent | BRAFi melanoma cell lines | [92] | |
| PD-L1 | ← (upregulates) | cJun | PD-L1 promoter binding. MAPK dependent | Lung carcinoma | [93] | |
| CTLA-4 | → (downregulates) | AP-1 | MAPK dependent | Inhibits T-cell activation | CD4+ T-cells or T-cell blasts | [102,103] |
| FOXP3 | → (downregulates) | cJun, cFos | JNK dependent | Maintains unresponsiveness of Tregs | Natural occurring Tregs | [110] |
| FOXP3 | ← (upregulates) | cJun, cFos | FOXP3 promoter binding. JNK dependent | Controls FOXP3 promoter activity | Sepsis model of Tregs | [111] |
| FOXP3 | ← (upregulates) | AP-1 | TGFb-induced FOXP3 promoter binding. MAPK dependent | Controls FOXP3 transcriptional activity | Tregs | [113] |
| FOXP3 | ← (downregulates) | BATF3 | FOXP3 promoter binding | Transcriptional suppressor of differentiation of Tregs | Tregs | [114] |
| ICOS, CTLA-4 | ← (upregulate) | JunB | IRF4 dependent | Loss of JunB in Tregs results in multi-organ autoimmunity | Tregs | [118] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers 2019, 11, 1037. https://doi.org/10.3390/cancers11071037
Atsaves V, Leventaki V, Rassidakis GZ, Claret FX. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers. 2019; 11(7):1037. https://doi.org/10.3390/cancers11071037
Chicago/Turabian StyleAtsaves, Vasileios, Vasiliki Leventaki, George Z. Rassidakis, and Francois X. Claret. 2019. "AP-1 Transcription Factors as Regulators of Immune Responses in Cancer" Cancers 11, no. 7: 1037. https://doi.org/10.3390/cancers11071037
APA StyleAtsaves, V., Leventaki, V., Rassidakis, G. Z., & Claret, F. X. (2019). AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers, 11(7), 1037. https://doi.org/10.3390/cancers11071037