Helping the Released Guardian: Drug Combinations for Supporting the Anticancer Activity of HDM2 (MDM2) Antagonists

 , , , ,
, , , ,
Abstract
1. Introduction
2. Limited Elimination of Cancer Cells by HDM2 Antagonists
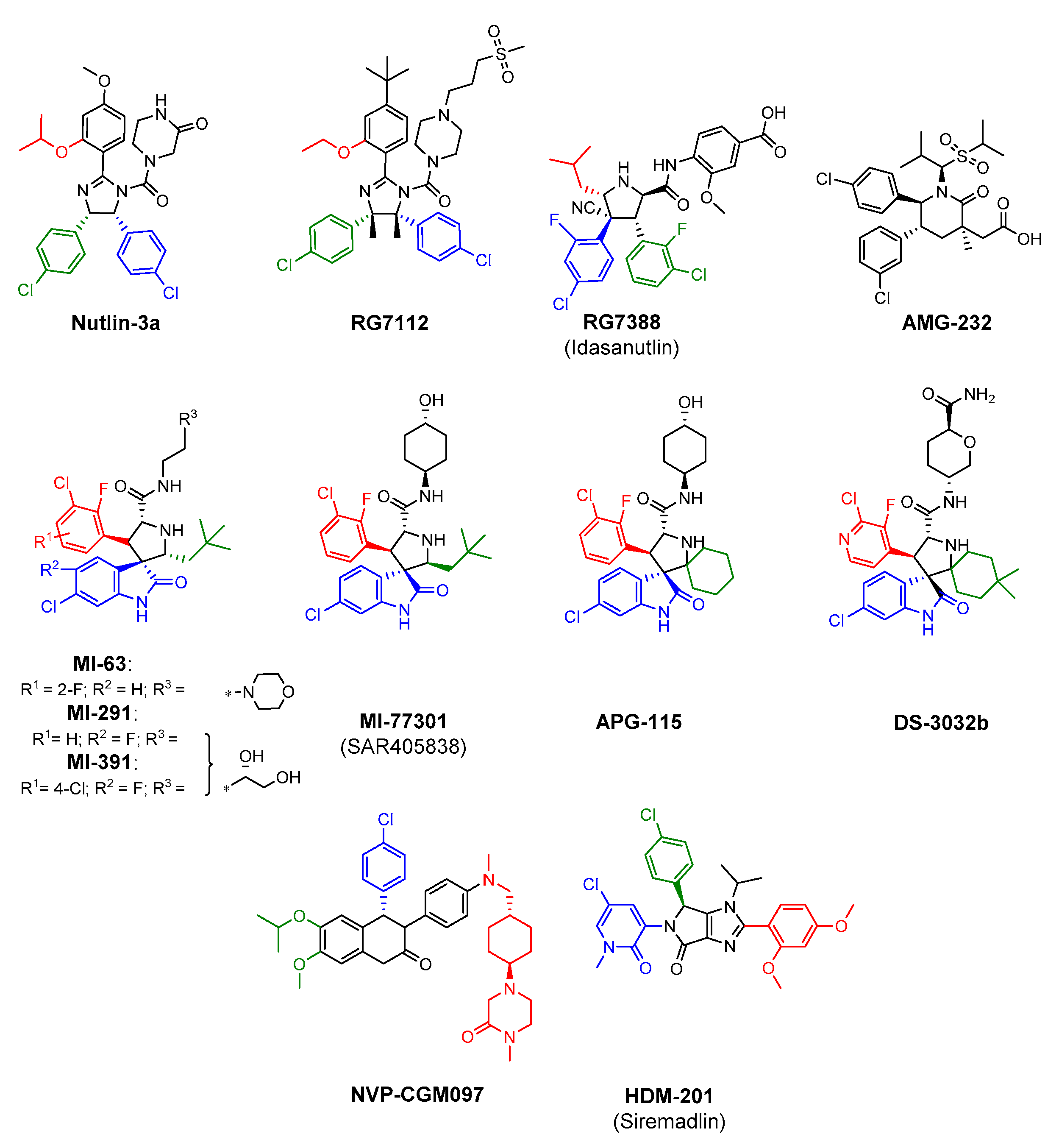
3. The Most Significant Representatives of HDM2 Antagonists
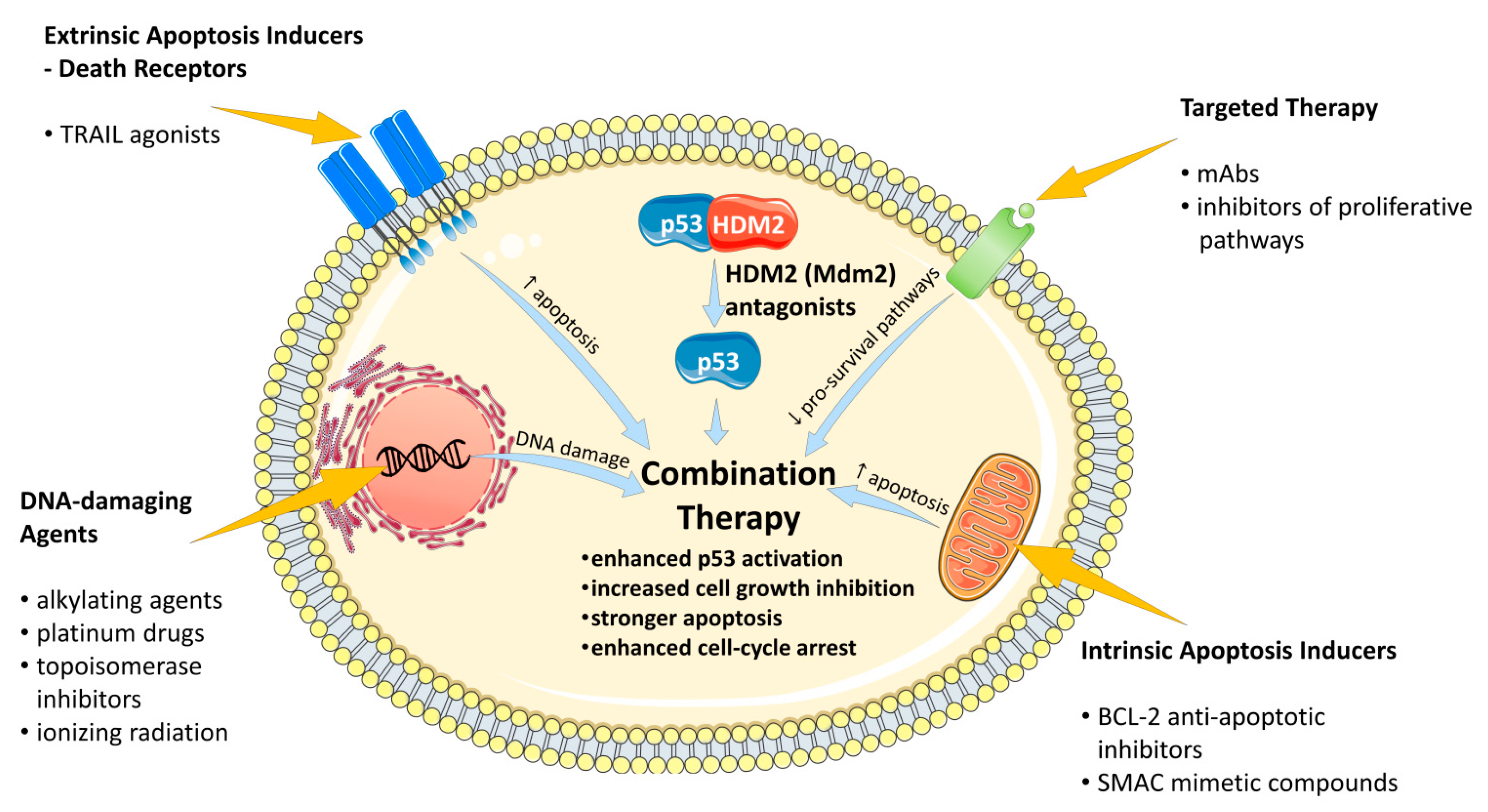
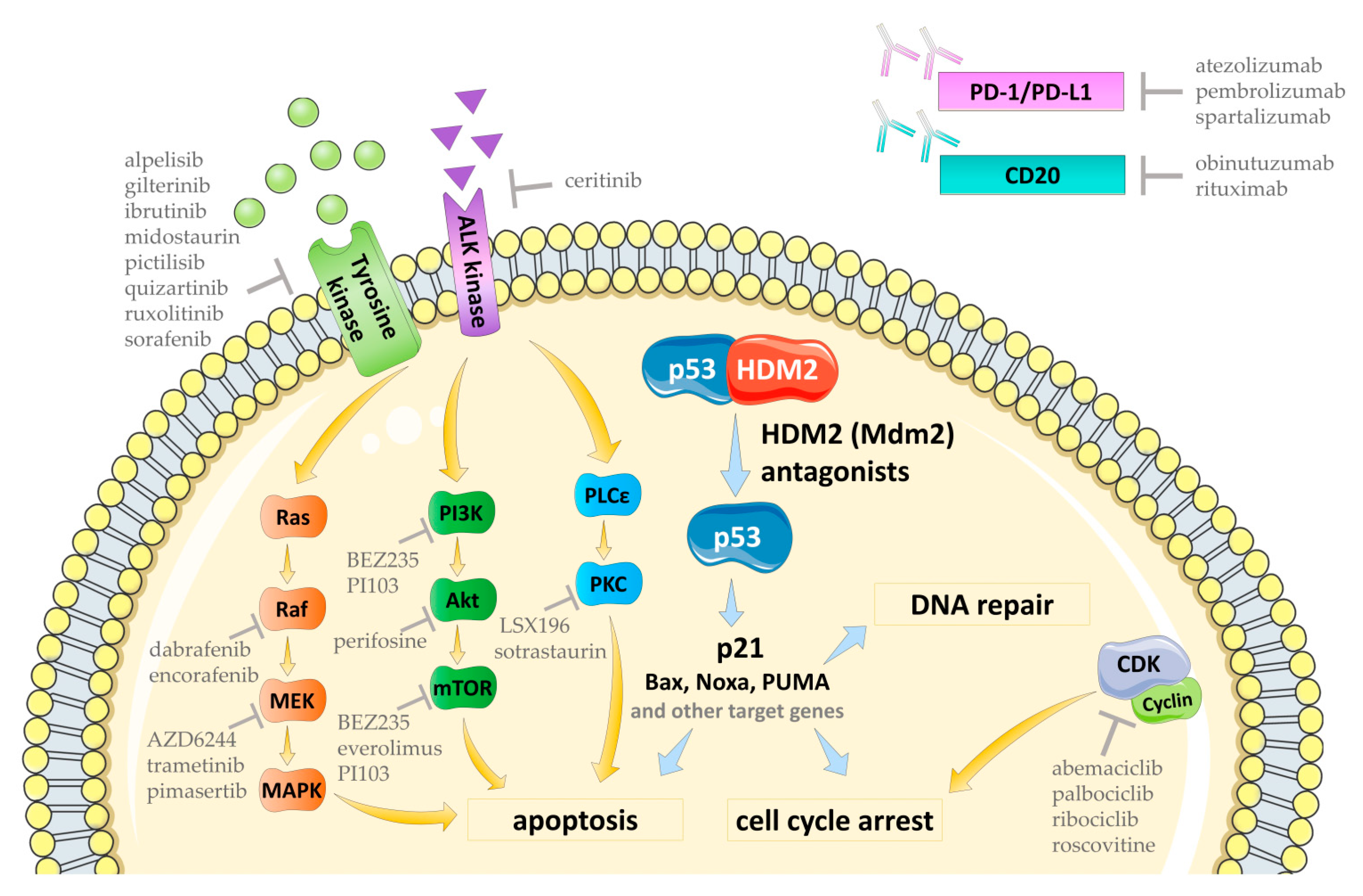
4. Drug Combinations for the Supporting of Anti-Cancer Activity of HDM2 Antagonists
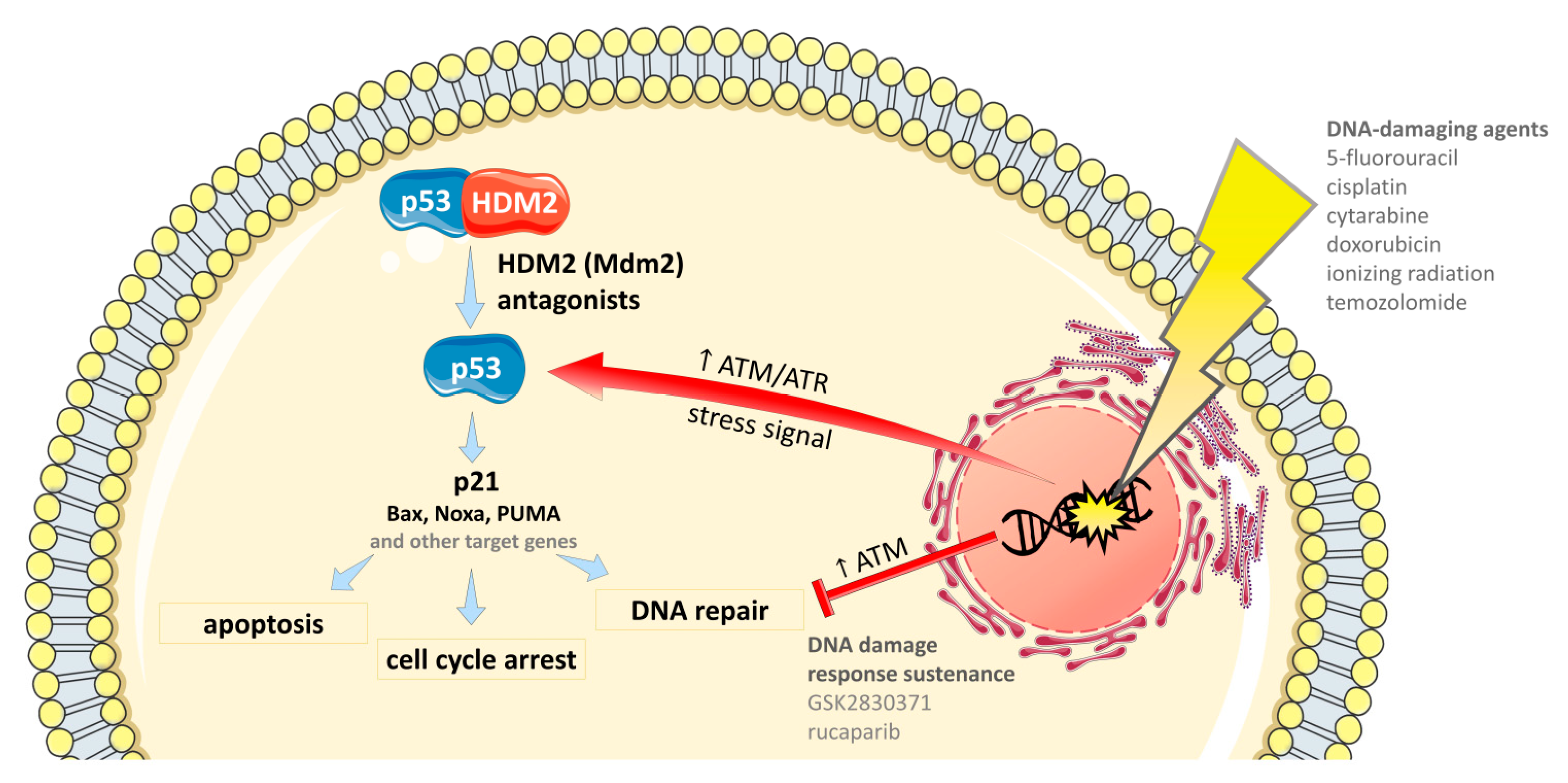
4.1. Combinations with DNA-Damaging Agents
4.1.1. Alkylating Agents
4.1.2. Platinum Complexes
4.1.3. Antimetabolites
4.1.4. Topoisomerase Inhibitors
4.1.5. Ionizing Radiation
4.2. Combinations with Drugs that Sustain DNA Damage Response
4.2.1. DNA Repair Inhibitors
4.2.2. Phosphatases
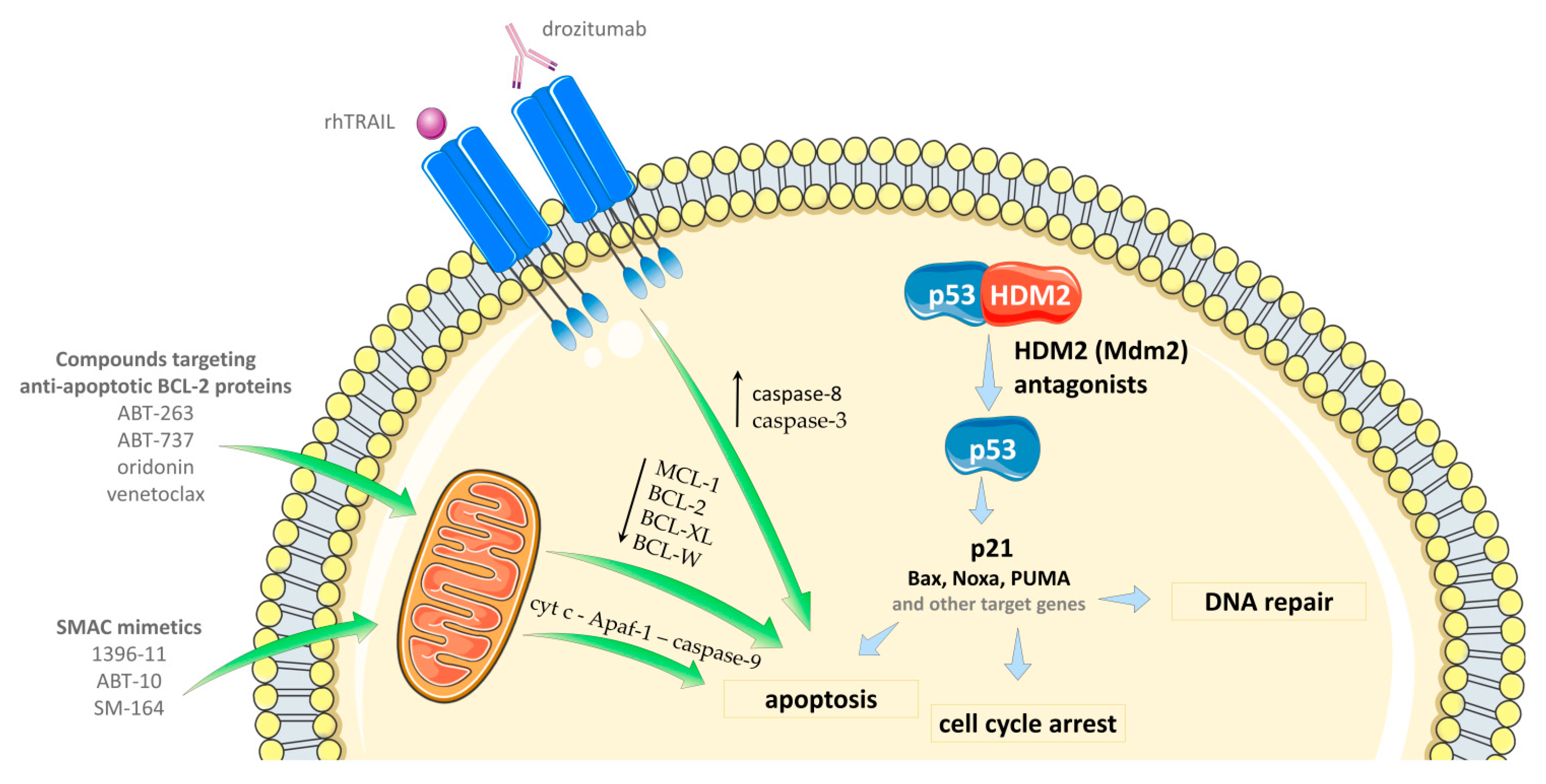
4.3. Combinations with Apoptosis-Inducing Agents
4.3.1. Selective BCL-2 Protein Inhibitors
4.3.2. Agents Downregulating the Level of Anti-Apoptotic BCL-2 Family Proteins
4.3.3. SMAC Mimetic Compounds
4.3.4. TRAIL Agonists
4.4. Combination with Therapy Selectively Targetting Pro-Survival Signaling Pathways
4.4.1. Inhibitors of Tyrosine Kinases
4.4.2. Ras/Raf/MEK/MAPK Pathway Inhibitors
4.4.3. PI3K/Akt/mTOR Pathway Inhibitors
4.4.4. CDK Inhibitors
4.4.5. PKC Inhibitors
4.5. The Combination with Anti-PD-/PD-L1 and Anti-CD20 Therapeutic Antibodies
4.6. The Combinations with Miscellaneous Anti-Cancer Agents
4.6.1. Proteasome Inhibitors
4.6.2. Histone Deacetylase Inhibitors
4.6.3. Antibiotics
4.6.4. Zinc
4.6.5. Heat Shock Protein Inhibitor
4.6.6. ATPase Inhibitors
4.6.7. Mitotic Inhibitors
4.6.8. Other Agents
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Catizone, A.N.; Good, C.R.; Alexander, K.A.; Berger, S.L.; Sammons, M.A. Comparison of genotoxic versus nongenotoxic stabilization of p53 provides insight into parallel stress-responsive transcriptional networks. Cell Cycle 2019, 18, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.; Vu, B.T.; Qing, W.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- París, R.; Henry, R.E.; Stephens, S.J.; McBryde, M.; Espinosa, J.M. Multiple p53-independent gene silencing mechanisms define the cellular response to p53 activation. Cell Cycle 2008, 7, 2427–2433. [Google Scholar] [CrossRef]
- Huang, B.; Deo, D.; Xia, M.; Vassilev, L.T. Pharmacologic p53 activation blocks cell cycle progression but fails to induce senescence in epithelial cancer cells. Mol. Cancer Res. 2009, 7, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; van Rikxoort, M.; Löschmann, N.; Voges, Y.; Breitling, R.; von Deimling, A.; Rödel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Shen, H.; Maki, C.G. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene 2011, 30, 4678–4686. [Google Scholar] [CrossRef]
- Wei, S.J.; Joseph, T.; Sim, A.Y.L.; Yurlova, L.; Zolghadr, K.; Lane, D.; Verma, C.; Ghadessy, F. In vitro selection of mutant HDM2 resistant to Nutlin inhibition. PLoS ONE 2013, 8, e62564. [Google Scholar] [CrossRef]
- Hoffman-Luca, C.G.; Ziazadeh, D.; McEachern, D.; Zhao, Y.; Sun, W.; Debussche, L.; Wang, S. Elucidation of Acquired Resistance to Bcl-2 and MDM2 Inhibitors in Acute Leukemia In Vitro and In Vivo. Clin. Cancer Res. 2015, 21, 2558–2568. [Google Scholar] [CrossRef]
- Jung, J.; Lee, J.S.; Dickson, M.A.; Schwartz, G.K.; Le Cesne, A.; Varga, A.; Bahleda, R.; Wagner, A.J.; Choy, E.; de Jonge, M.J.; et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat. Commun. 2016, 7, 12609. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.J.; Esfandiari, A.; Liu, J.; Lu, X.; Hutton, C.; Jackson, J.; Bennaceur, K.; Xu, Q.; Makimanejavali, A.R.; Del Bello, F.; et al. TP53 mutant MDM2-amplified cell lines selected for resistance to MDM2-p53 binding antagonists retain sensitivity to ionizing radiation. Oncotarget 2016, 7, 46203–46218. [Google Scholar] [CrossRef] [PubMed]
- Chapeau, E.A.; Gembarska, A.; Durand, E.Y.; Mandon, E.; Estadieu, C.; Romanet, V.; Wiesmann, M.; Tiedt, R.; Lehar, J.; de Weck, A.; et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf−/− mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, 3151–3156. [Google Scholar] [CrossRef] [PubMed]
- Skalniak, L.; Kocik, J.; Polak, J.; Skalniak, A.; Rak, M.; Wolnicka-Glubisz, A.; Holak, T.A. Prolonged Idasanutlin (RG7388) Treatment Leads to the Generation of p53-Mutated Cells. Cancers 2018, 10, 396. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-E.; Esfandiari, A.; Ho, Y.-H.; Wang, N.; Mahdi, A.K.; Aptullahoglu, E.; Lovat, P.; Lunec, J. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br. J. Cancer 2018, 118, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Jeay, S.; Gaulis, S.; Ferretti, S.; Bitter, H.; Ito, M.; Valat, T.; Murakami, M.; Ruetz, S.; Guthy, D.A.; Rynn, C.; et al. A distinct p53 target gene set predicts for response to the selective p53-HDM2 inhibitor NVP-CGM097. Elife 2015, 4, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.D.; Xuan, Y.; Broude, E.V.; Zhu, H.; Schott, B.; Fang, J.; Roninson, I.B. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 1999, 18, 4808–4818. [Google Scholar] [CrossRef]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent initiation: A mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell 2008, 30, 277–289. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef]
- Zhang, X.-P.; Liu, F.; Wang, W. Coordination between cell cycle progression and cell fate decision by the p53 and E2F1 pathways in response to DNA damage. J. Biol. Chem. 2010, 285, 31571–31580. [Google Scholar] [CrossRef] [PubMed]
- Hat, B.; Kochańczyk, M.; Bogdał, M.N.; Lipniacki, T. Feedbacks, Bifurcations, and Cell Fate Decision-Making in the p53 System. PLoS Comput. Biol. 2016, 12, e1004787. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [PubMed]
- Puca, R.; Nardinocchi, L.; Givol, D.; D’Orazi, G. Regulation of p53 activity by HIPK2: Molecular mechanisms and therapeutical implications in human cancer cells. Oncogene 2010, 29, 4378–4387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, F.; Cheng, Z.; Wang, W. Cell fate decision mediated by p53 pulses. Proc. Natl. Acad. Sci. USA 2009, 106, 12245–12250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-P.; Liu, F.; Wang, W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef] [PubMed]
- Kracikova, M.; Akiri, G.; George, A.; Sachidanandam, R.; Aaronson, S.A. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013, 20, 576–588. [Google Scholar] [CrossRef]
- Krenning, L.; Feringa, F.M.; Shaltiel, I.A.; van den Berg, J.; Medema, R.H. Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol. Cell 2014, 55, 59–72. [Google Scholar] [CrossRef]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 133. [Google Scholar] [CrossRef]
- Skalniak, L.; Surmiak, E.; Holak, T.A. A therapeutic patent overview of MDM2/X-targeted therapies (2014-2018). Expert Opin. Ther. Pat. 2019, 29, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Reuther, C.; Heinzle, V.; Nölting, S.; Herterich, S.; Hahner, S.; Halilovic, E.; Jeay, S.; Wuerthner, J.U.; Aristizabal Prada, E.T.; Spöttl, G.; et al. The HDM2 (MDM2) Inhibitor NVP-CGM097 Inhibits Tumor Cell Proliferation and Shows Additive Effects with 5-Fluorouracil on the p53-p21-Rb-E2F1 Cascade in the p53wild type Neuroendocrine Tumor Cell Line GOT1. Neuroendocrinology 2018, 106, 1–19. [Google Scholar] [CrossRef]
- Coll-Mulet, L.; Iglesias-Serret, D.; Santidrián, A.F.; Cosialls, A.M.; de Frias, M.; Castaño, E.; Campàs, C.; Barragán, M.; de Sevilla, A.F.; Domingo, A.; et al. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B-cell chronic lymphocytic leukemia cells. Blood 2006, 107, 4109–4114. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Rousseau, R.F.; Middleton, S.A.; Nichols, G.L.; Newell, D.R.; Lunec, J.; Tweddle, D.A. Pre-clinical evaluation of the MDM2-p53 antagonist RG7388 alone and in combination with chemotherapy in neuroblastoma. Oncotarget 2015, 6, 10207–10221. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.Y.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Tonsing-Carter, E.; Bailey, B.J.; Saadatzadeh, M.R.; Ding, J.; Wang, H.; Sinn, A.L.; Peterman, K.M.; Spragins, T.K.; Silver, J.M.; Sprouse, A.A.; et al. Potentiation of Carboplatin-Mediated DNA Damage by the Mdm2 Modulator Nutlin-3a in a Humanized Orthotopic Breast-to-Lung Metastatic Model. Mol. Cancer Ther. 2015, 14, 2850–2863. [Google Scholar] [CrossRef] [PubMed]
- Zanjirband, M.; Edmondson, R.J.; Lunec, J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget 2016, 7, 40115–40134. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Aboukameel, A.; Banerjee, S.; Wang, Z.; Mohammad, M.; Wu, J.; Wang, S.; Yang, D.; Philip, P.A.; Sarkar, F.H.; et al. MDM2 inhibitor MI-319 in combination with cisplatin is an effective treatment for pancreatic cancer independent of p53 function. Eur. J. Cancer 2010, 46, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Voon, Y.-L.; Ahmad, M.; Wong, P.; Husaini, R.; Ng, W.T.; Leong, C.-O.; Lane, D.P.; Khoo, A.S.-B. Nutlin-3 sensitizes nasopharyngeal carcinoma cells to cisplatin-induced cytotoxicity. Oncol. Rep. 2015, 34, 1692–1700. [Google Scholar] [CrossRef]
- Deben, C.; Wouters, A.; Op de Beeck, K.; van Den Bossche, J.; Jacobs, J.; Zwaenepoel, K.; Peeters, M.; Van Meerbeeck, J.; Lardon, F.; Rolfo, C.; et al. The MDM2-inhibitor Nutlin-3 synergizes with cisplatin to induce p53 dependent tumor cell apoptosis in non-small cell lung cancer. Oncotarget 2015, 6, 22666–22679. [Google Scholar] [CrossRef]
- Carrillo, A.M.; Hicks, M.; Khabele, D.; Eischen, C.M. Pharmacologically Increasing Mdm2 Inhibits DNA Repair and Cooperates with Genotoxic Agents to Kill p53-Inactivated Ovarian Cancer Cells. Mol. Cancer Res. 2015, 13, 1197–1205. [Google Scholar] [CrossRef]
- Ohnstad, H.O.; Paulsen, E.B.; Noordhuis, P.; Berg, M.; Lothe, R.A.; Vassilev, L.T.; Myklebost, O. MDM2 antagonist Nutlin-3a potentiates antitumour activity of cytotoxic drugs in sarcoma cell lines. BMC Cancer 2011, 11, 211. [Google Scholar] [CrossRef]
- Barbieri, E.; Mehta, P.; Chen, Z.; Zhang, L.; Slack, A.; Berg, S.; Shohet, J.M. MDM2 inhibition sensitizes neuroblastoma to chemotherapy-induced apoptotic cell death. Mol. Cancer Ther. 2006, 5, 2358–2365. [Google Scholar] [CrossRef]
- Seipel, K.; Marques, M.A.T.; Sidler, C.; Mueller, B.U.; Pabst, T. MDM2- and FLT3-inhibitors in the treatment of FLT3 -ITD acute myeloid leukemia, specificity and efficacy of NVP-HDM201 and midostaurin. Haematologica 2018, 103, 1862–1872. [Google Scholar] [CrossRef]
- Drakos, E.; Singh, R.R.; Rassidakis, G.Z.; Schlette, E.; Li, J.; Claret, F.X.; Ford, R.J.; Vega, F.; Medeiros, L.J. Activation of the p53 pathway by the MDM2 inhibitor nutlin-3a overcomes BCL2 overexpression in a preclinical model of diffuse large B-cell lymphoma associated with t(14;18) (q32;q21). Leukemia 2011, 25, 856–867. [Google Scholar] [CrossRef]
- Conradt, L.; Henrich, A.; Wirth, M.; Reichert, M.; Lesina, M.; Algül, H.; Schmid, R.M.; Krämer, O.H.; Saur, D.; Schneider, G. Mdm2 inhibitors synergize with topoisomerase II inhibitors to induce p53-independent pancreatic cancer cell death. Int. J. Cancer 2013, 132, 2248–2257. [Google Scholar] [CrossRef]
- Zheng, M.; Yang, J.; Xu, X.; Sebolt, J.T.; Wang, S.; Sun, Y. Efficacy of MDM2 inhibitor MI-219 against lung cancer cells alone or in combination with MDM2 knockdown, a XIAP inhibitor or etoposide. Anticancer Res. 2010, 30, 3321–3331. [Google Scholar]
- Kojima, K.; Konopleva, M.; McQueen, T.; Brien, S.O.; Plunkett, W.; Andreeff, M.; O’Brien, S.; Plunkett, W.; Andreeff, M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood 2006, 108, 993–1000. [Google Scholar] [CrossRef]
- Jones, R.J.; Baladandayuthapani, V.; Neelapu, S.; Fayad, L.E.; Romaguera, J.E.; Wang, M.; Sharma, R.; Yang, D.; Orlowski, R.Z. HDM-2 inhibition suppresses expression of ribonucleotide reductase subunit M2, and synergistically enhances gemcitabine-induced cytotoxicity in mantle cell lymphoma. Blood 2011, 118, 4140–4149. [Google Scholar] [CrossRef]
- Prabakaran, P.J.; Javaid, A.M.; Swick, A.D.; Werner, L.R.; Nickel, K.P.; Sampene, E.; Hu, R.; Ong, I.M.; Bruce, J.Y.; Hartig, G.K.; et al. Radiosensitization of Adenoid Cystic Carcinoma with MDM2 Inhibition. Clin. Cancer Res. 2017, 23, 6044–6053. [Google Scholar] [CrossRef]
- Phelps, D.; Bondra, K.; Seum, S.; Chronowski, C.; Leasure, J.; Kurmasheva, R.T.; Middleton, S.; Wang, D.; Mo, X.; Houghton, P.J. Inhibition of MDM2 by RG7388 confers hypersensitivity to X-radiation in xenograft models of childhood sarcoma. Pediatr. Blood Cancer 2015, 62, 1345–1352. [Google Scholar] [CrossRef]
- Arya, A.K.; El-Fert, A.; Devling, T.; Eccles, R.M.; Aslam, M.A.; Rubbi, C.P.; Vlatković, N.; Fenwick, J.; Lloyd, B.H.; Sibson, D.R.; et al. Nutlin-3, the small-molecule inhibitor of MDM2, promotes senescence and radiosensitises laryngeal carcinoma cells harbouring wild-type p53. Br. J. Cancer 2010, 103, 186–195. [Google Scholar] [CrossRef]
- Villalonga-Planells, R.; Coll-Mulet, L.; Martínez-Soler, F.; Castaño, E.; Acebes, J.-J.; Giménez-Bonafé, P.; Gil, J.; Tortosa, A. Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS ONE 2011, 6, e18588. [Google Scholar] [CrossRef]
- Azmi, A.S.; Banerjee, S.; Ali, S.; Wang, Z.; Bao, B.; Beck, F.W.J.; Maitah, M.; Choi, M.; Shields, T.F.; Philip, P.A.; et al. Network modeling of MDM2 inhibitor-oxaliplatin combination reveals biological synergy in wt-p53 solid tumors. Oncotarget 2011, 2, 378–392. [Google Scholar] [CrossRef]
- Chen, L.; Pastorino, F.; Berry, P.; Bonner, J.; Kirk, C.; Wood, K.M.; Thomas, H.D.; Zhao, Y.; Daga, A.; Veal, G.J.; et al. Preclinical evaluation of the first intravenous small molecule MDM2 antagonist alone and in combination with temozolomide in neuroblastoma. Int. J. Cancer 2019, 144, 3146–3159. [Google Scholar] [CrossRef]
- Obrador-Hevia, A.; Martinez-Font, E.; Felipe-Abrio, I.; Calabuig-Fariñas, S.; Serra-Sitjar, M.; López-Guerrero, J.A.; Ramos, R.; Alemany, R.; Martín-Broto, J. RG7112, a small-molecule inhibitor of MDM2, enhances trabectedin response in soft tissue sarcomas. Cancer Invest. 2015, 33, 440–450. [Google Scholar] [CrossRef]
- Stolte, B.; Iniguez, A.B.; Dharia, N.V.; Robichaud, A.L.; Conway, A.S.; Morgan, A.M.; Alexe, G.; Schauer, N.J.; Liu, X.; Bird, G.H.; et al. Genome-scale CRISPR-Cas9 screen identifies druggable dependencies in TP53 wild-type Ewing sarcoma. J. Exp. Med. 2018, 215, 2137–2155. [Google Scholar] [CrossRef]
- Sriraman, A.; Radovanovic, M.; Wienken, M.; Najafova, Z.; Li, Y.; Dobbelstein, M. Cooperation of Nutlin-3a and a Wip1 inhibitor to induce p53 activity. Oncotarget 2016, 7, 31623–31638. [Google Scholar] [CrossRef]
- Zanjirband, M.; Curtin, N.; Edmondson, R.J.; Lunec, J. Combination treatment with rucaparib (Rubraca) and MDM2 inhibitors, Nutlin-3 and RG7388, has synergistic and dose reduction potential in ovarian cancer. Oncotarget 2017, 8, 69779–69796. [Google Scholar] [CrossRef]
- Kojima, K.; Konopleva, M.; Samudio, I.J.; Schober, W.D.; Bornmann, W.G.; Andreeff, M. Concomitant inhibition of MDM2 and Bcl-2 protein function synergistically induce mitochondrial apoptosis in AML. Cell Cycle 2006, 5, 2778–2786. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Mak, D.H.; Ruvolo, V.R.; Schober, W.; McQueen, T.; Cortes, J.; Kantarjian, H.M.; Champlin, R.E.; Konopleva, M.; et al. Synergistic effects of p53 activation via MDM2 inhibition in combination with inhibition of Bcl-2 or Bcr-Abl in CD34+ proliferating and quiescent chronic myeloid leukemia blast crisis cells. Oncotarget 2015, 6, 30487–30499. [Google Scholar] [CrossRef]
- Gu, D.; Wang, S.; Kuiatse, I.; Wang, H.; He, J.; Dai, Y.; Jones, R.J.; Bjorklund, C.C.; Yang, J.; Grant, S.; et al. Inhibition of the MDM2 E3 Ligase induces apoptosis and autophagy in wild-type and mutant p53 models of multiple myeloma, and acts synergistically with ABT-737. PLoS ONE 2014, 9, e103015. [Google Scholar] [CrossRef]
- Wang, X.-H.; Zhang, S.-F.; Bao, J.-T.; Liu, F.-Y. Oridonin synergizes with Nutlin-3 in osteosarcoma cells by modulating the levels of multiple Bcl-2 family proteins. Tumour Biol. 2017, 39, 1010428317701638. [Google Scholar] [CrossRef]
- Lehmann, C.; Friess, T.; Birzele, F.; Kiialainen, A.; Dangl, M. Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J. Hematol. Oncol. 2016, 9, 50. [Google Scholar] [CrossRef]
- Van Goethem, A.; Yigit, N.; Moreno-Smith, M.; Vasudevan, S.A.; Barbieri, E.; Speleman, F.; Shohet, J.; Vandesompele, J.; Van Maerken, T. Dual targeting of MDM2 and BCL2 as a therapeutic strategy in neuroblastoma. Oncotarget 2017, 8, 57047–57057. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, D.H.; Schober, W.D.; Koller, E.; Pinilla, C.; Vassilev, L.T.; Reed, J.C.; Andreeff, M. Simultaneous activation of p53 and inhibition of XIAP enhance the activation of apoptosis signaling pathways in AML. Blood 2010, 115, 306–314. [Google Scholar] [CrossRef]
- Meijer, A.; Kruyt, F.A.E.; Van Der Zee, A.G.J.; Hollema, H.; Le, P.; Ten Hoor, K.A.; Groothuis, G.M.M.; Quax, W.J.; De Vries, E.G.E.; De Jong, S. Nutlin-3 preferentially sensitises wild-type p53-expressing cancer cells to DR5-selective TRAIL over rhTRAIL. Br. J. Cancer 2013, 109, 2685–2695. [Google Scholar] [CrossRef]
- Urso, L.; Cavallari, I.; Silic-Benussi, M.; Biasini, L.; Zago, G.; Calabrese, F.; Conte, P.F.; Ciminale, V.; Pasello, G. Synergistic targeting of malignant pleural mesothelioma cells by MDM2 inhibitors and TRAIL agonists. Oncotarget 2017, 8, 44232–44241. [Google Scholar] [CrossRef][Green Version]
- Wang, H.Q.; Halilovic, E.; Li, X.; Liang, J.; Cao, Y.; Rakiec, D.P.; Ruddy, D.A.; Jeay, S.; Wuerthner, J.U.; Timple, N.; et al. Combined ALK and MDM2 inhibition increases antitumor activity and overcomes resistance in human ALK mutant neuroblastoma cell lines and xenograft models. Elife 2017, 6, 1–19. [Google Scholar] [CrossRef]
- Zauli, G.; Voltan, R.; Bosco, R.; Melloni, E.; Marmiroli, S.; Rigolin, G.M.; Cuneo, A.; Secchiero, P. Dasatinib plus nutlin-3 shows synergistic antileukemic activity in both p53wild-typeand p53mutated B chronic lymphocytic leukemias by inhibiting the Akt pathway. Clin. Cancer Res. 2011, 17, 762–770. [Google Scholar] [CrossRef]
- Trino, S.; Iacobucci, I.; Erriquez, D.; Laurenzana, I.; De Luca, L.; Ferrari, A.; Di Rorà, A.G.L.; Papayannidis, C.; Derenzini, E.; Simonetti, G.; et al. Targeting the p53-MDM2 interaction by the small-molecule MDM2 antagonist Nutlin-3a: A new challenged target therapy in adult Philadelphia positive acute lymphoblastic leukemia patients. Oncotarget 2016, 7, 12951–12961. [Google Scholar] [CrossRef]
- Voltan, R.; Rimondi, E.; Melloni, E.; Rigolin, G.M.; Casciano, F.; Arcidiacono, M.V.; Celeghini, C.; Cuneo, A.; Zauli, G.; Secchiero, P. Ibrutinib synergizes with MDM-2 inhibitors in promoting cytotoxicity in B chronic lymphocytic leukemia. Oncotarget 2016, 7, 70623–70638. [Google Scholar] [CrossRef][Green Version]
- Weisberg, E.; Halilovic, E.; Cooke, V.G.; Nonami, A.; Ren, T.; Sanda, T.; Simkin, I.; Yuan, J.; Antonakos, B.; Barys, L.; et al. Inhibition of Wild-Type p53-Expressing AML by the Novel Small Molecule HDM2 Inhibitor CGM097. Mol. Cancer Ther. 2015, 14, 2249–2259. [Google Scholar] [CrossRef]
- Zauli, G.; Celeghini, C.; Melloni, E.; Voltan, R.; Ongari, M.; Tiribelli, M.; di Iasio, M.G.; Lanza, F.; Secchiero, P. The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica 2012, 97, 1722–1730. [Google Scholar] [CrossRef]
- Vatsyayan, R.; Singhal, J.; Nagaprashantha, L.D.; Awasthi, S.; Singhal, S.S. Nutlin-3 enhances sorafenib efficacy in renal cell carcinoma. Mol. Carcinog. 2013, 52, 39–48. [Google Scholar] [CrossRef]
- McIntosh, J.; Huang, X.; Yan, X.; Lo, M.-C.; Ye, Q.; Houze, J.B.; Mihalic, J.; Oliner, J.D.; Medina, J.C.; Yu, M.; et al. Discovery of AMG 232, a Potent, Selective, and Orally Bioavailable MDM2-p53 Inhibitor in Clinical Development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar]
- Zhang, W.; Konopleva, M.; Burks, J.K.; Dywer, K.C.; Schober, W.D.; Yang, J.-Y.; McQueen, T.J.; Hung, M.-C.; Andreeff, M. Blockade of MEK and MDM2 synergistically induces apoptosis in acute myeloid leukemia via BH3-only proteins Puma and Bim. Cancer Res. 2010, 70, 2424–2434. [Google Scholar] [CrossRef]
- Moschos, S.J.; Sandhu, S.K.; Lewis, K.D.; Sullivan, R.J.; Johnson, D.B.; Zhang, Y.; Rasmussen, E.; Henary, H.A.; Long, G.V. Phase 1 study of the p53-MDM2 inhibitor AMG 232 combined with trametinib plus dabrafenib or trametinib in patients (Pts) with TP53 wild type (TP53WT) metastatic cutaneous melanoma (MCM). J. Clin. Oncol. 2017, 35, 2575. [Google Scholar] [CrossRef]
- Saiki, A.Y.; Caenepeel, S.; Yu, D.; Lofgren, J.A.; Osgood, T.; Robertson, R.; Canon, J.; Su, C.; Jones, A.; Zhao, X.; et al. MDM2 antagonists synergize broadly and robustly with compounds targeting fundamental oncogenic signaling pathways. Oncotarget 2014, 5, 2030–2043. [Google Scholar] [CrossRef]
- Wang, H.Q.; Zubrowski, M.; Emerson, E.; Pradhan, E.; Jeay, S.; Wiesmann, M.; Caponigro, G.; Wuerthner, J.; Schlegel, R.; Cao, Z.A.; et al. Abstract 5466: The Mdm2 inhibitor, NVP-CGM097, in combination with the BRAF inhibitor NVP-LGX818 elicits synergistic antitumor effects in melanoma. In Proceedings of the 2014 Annual Meeting of the American Association for Cancer Research, Experimental and Molecular Therapeutics, San Diego, CA, USA, 5–9 April 2014; Volume 74, p. 5466. [Google Scholar]
- de Weger, V.A.; de Jonge, M.; Langenberg, M.H.G.G.; Schellens, J.H.M.M.; Lolkema, M.; Varga, A.; Demers, B.; Thomas, K.; Hsu, K.; Tuffal, G.; et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 286–293. [Google Scholar] [CrossRef]
- Wu, C.-E.; Koay, T.S.; Esfandiari, A.; Ho, Y.-H.; Lovat, P.; Lunec, J. ATM Dependent DUSP6 Modulation of p53 Involved in Synergistic Targeting of MAPK and p53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers 2018, 11, 3. [Google Scholar] [CrossRef]
- Laroche, A.; Chaire, V.; Algeo, M.-P.; Karanian, M.; Fourneaux, B.; Italiano, A. MDM2 antagonists synergize with PI3K/mTOR inhibition in well-differentiated/dedifferentiated liposarcomas. Oncotarget 2017, 8, 53968–53977. [Google Scholar] [CrossRef]
- Zhu, N.; Gu, L.; Li, F.; Zhou, M. Inhibition of the Akt/survivin pathway synergizes the antileukemia effect of nutlin-3 in acute lymphoblastic leukemia cells. Mol. Cancer Ther. 2008, 7, 1101–1109. [Google Scholar] [CrossRef]
- Kojima, K.; Shimanuki, M.; Shikami, M.; Samudio, I.J.; Ruvolo, V.; Corn, P.; Hanaoka, N.; Konopleva, M.; Andreeff, M.; Nakakuma, H. The dual PI3 kinase/mTOR inhibitor PI-103 prevents p53 induction by Mdm2 inhibition but enhances p53-mediated mitochondrial apoptosis in p53 wild-type AML. Leukemia 2008, 22, 1728–1736. [Google Scholar] [CrossRef]
- Schmeits, P.C.J.; Hermans, M.H.A.; Van Geest-daalderop, J.H.H.; Poodt, J.; Nolting, P.R.W.D.S.; Conemans, J.M.H. Perifosine plus nutlin-3 combination shows a synergistic anti-leukaemic activity. Br. J. Haematol. 2009, 3, 957–961. [Google Scholar]
- Sriraman, A.; Dickmanns, A.; Najafova, Z.; Johnsen, S.A.; Dobbelstein, M. CDK4 inhibition diminishes p53 activation by MDM2 antagonists. Cell Death Dis. 2018, 9, 918. [Google Scholar] [CrossRef]
- Laroche-Clary, A.; Chaire, V.; Algeo, M.-P.; Derieppe, M.-A.; Loarer, F.L.; Italiano, A. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J. Hematol. Oncol. 2017, 10, 123. [Google Scholar] [CrossRef]
- Ribas, J.; Boix, J.; Meijer, L. (R)-roscovitine (CYC202, Seliciclib) sensitizes SH-SY5Y neuroblastoma cells to nutlin-3-induced apoptosis. Exp. Cell Res. 2006, 312, 2394–2400. [Google Scholar] [CrossRef]
- Cheok, C.F.; Dey, A.; Lane, D.P. Cyclin-dependent kinase inhibitors sensitize tumor cells to nutlin-induced apoptosis: A potent drug combination. Mol. Cancer Res. 2007, 5, 1133–1145. [Google Scholar] [CrossRef]
- Carita, G.; Frisch-Dit-Leitz, E.; Dahmani, A.; Raymondie, C.; Cassoux, N.; Piperno-Neumann, S.; Némati, F.; Laurent, C.; De Koning, L.; Halilovic, E.; et al. Dual inhibition of protein kinase C and p53-MDM2 or PKC and mTORC1 are novel efficient therapeutic approaches for uveal melanoma. Oncotarget 2016, 7, 33542–33556. [Google Scholar] [CrossRef]
- Jak, M.; Reits, E.A.; Kallemeijn, W.W.; Tromp, J.M.; Klein, C.; Eldering, E. CD40 stimulation sensitizes CLL cells to lysosomal cell death induction by type II anti-CD20 monoclonal antibody GA101. Blood 2011, 118, 5178–5189. [Google Scholar] [CrossRef][Green Version]
- Herting, F.; Herter, S.; Friess, T.; Muth, G.; Bacac, M.; Sulcova, J.; Umana, P.; Dangl, M.; Klein, C. Antitumour activity of the glycoengineered type II anti-CD20 antibody obinutuzumab (GA101) in combination with the MDM2-selective antagonist idasanutlin (RG7388). Eur. J. Haematol. 2016, 97, 461–470. [Google Scholar] [CrossRef]
- Pishas, K.I.; Neuhaus, S.J.; Clayer, M.T.; Adwal, A.; Brown, M.P.; Evdokiou, A.; Callen, D.F.; Neilsen, P.M. Pre-activation of the p53 pathway through Nutlin-3a sensitises sarcomas to drozitumab therapy. Oncol. Rep. 2013, 30, 471–477. [Google Scholar] [CrossRef]
- Ooi, M.G.; Hayden, P.J.; Kotoula, V.; McMillin, D.W.; Charalambous, E.; Daskalaki, E.; Raje, N.S.; Munshi, N.C.; Chauhan, D.; Hideshima, T.; et al. Interactions of the Hdm2/p53 and proteasome pathways may enhance the antitumor activity of bortezomib. Clin. Cancer Res. 2009, 15, 7153–7160. [Google Scholar] [CrossRef]
- Saha, M.N.; Jiang, H.; Jayakar, J.; Reece, D.; Branch, D.R.; Chang, H. MDM2 antagonist nutlin plus proteasome inhibitor velcade combination displays a synergistic anti-myeloma activity. Cancer Biol. Ther. 2010, 9, 936–944. [Google Scholar] [CrossRef]
- Tabe, Y.; Sebasigari, D.; Jin, L.; Rudelius, M.; Davies-Hill, T.; Miyake, K.; Miida, T.; Pittaluga, S.; Raffeld, M. MDM2 antagonist nutlin-3 displays antiproliferative and proapoptotic activity in mantle cell lymphoma. Clin. Cancer Res. 2009, 15, 933–942. [Google Scholar] [CrossRef]
- Chen, H.; Xue, L.; Huang, H.; Wang, H.; Zhang, X.; Zhu, W.; Wang, Z.; Wang, Z.; Wu, H. Synergistic effect of Nutlin-3 combined with MG-132 on schwannoma cells through restoration of merlin and p53 tumour suppressors. EBioMedicine 2018, 36, 252–265. [Google Scholar] [CrossRef]
- Borthakur, G.; Duvvuri, S.; Samudio, I.J.; Kojima, K.; Konopleva, M.; Andreeff, M. Synergistic Activation of p53 Family of Proteins by the Combination of Nutlin 3a and SAHA in Acute Myelogenous Leukemia. Blood 2009, 114, 1738. [Google Scholar]
- McCormack, E.; Haaland, I.; Venås, G.; Forthun, R.B.; Huseby, S.; Gausdal, G.; Knappskog, S.; Micklem, D.R.; Lorens, J.B.; Bruserud, O.; et al. Synergistic induction of p53 mediated apoptosis by valproic acid and nutlin-3 in acute myeloid leukemia. Leukemia 2012, 26, 910–917. [Google Scholar] [CrossRef]
- Miyachi, M.; Kakazu, N.; Yagyu, S.; Katsumi, Y.; Tsubai-Shimizu, S.; Kikuchi, K.; Tsuchiya, K.; Iehara, T.; Hosoi, H. Restoration of p53 pathway by nutlin-3 induces cell cycle arrest and apoptosis in human rhabdomyosarcoma cells. Clin. Cancer Res. 2009, 15, 4077–4084. [Google Scholar] [CrossRef]
- Zajkowicz, A.; Gdowicz-Kłosok, A.; Krześniak, M.; Ścieglińska, D.; Rusin, M. Actinomycin D and nutlin-3a synergistically promote phosphorylation of p53 on serine 46 in cancer cell lines of different origin. Cell. Signal. 2015, 27, 1677–1687. [Google Scholar] [CrossRef]
- Zaman, S.; Yu, X.; Bencivenga, A.F.; Blanden, A.R.; Liu, Y.; Withers, T.; Na, B.; Blayney, A.J.; Gilleran, J.; Boothman, D.A.; et al. Combinatorial therapy of Zinc metallochaperones with mutant p53 reactivation and diminished copper binding. Mol. Cancer Ther. 2019. [Google Scholar] [CrossRef]
- Haaland, I.; Opsahl, J.A.; Berven, F.S.; Reikvam, H.; Fredly, H.K.; Haugse, R.; Thiede, B.; McCormack, E.; Lain, S.; Bruserud, O.; et al. Molecular mechanisms of nutlin-3 involve acetylation of p53, histones and heat shock proteins in acute myeloid leukemia. Mol. Cancer 2014, 13, 116. [Google Scholar] [CrossRef]
- Schneider, L.S.; Ulrich, M.; Lehr, T.; Menche, D.; Müller, R.; von Schwarzenberg, K. MDM2 antagonist nutlin-3a sensitizes tumors to V-ATPase inhibition. Mol. Oncol. 2016, 10, 1054–1062. [Google Scholar] [CrossRef]
- Richmond, J.; Carol, H.; Evans, K.; High, L.; Mendomo, A.; Robbins, A.; Meyer, C.; Venn, N.C.; Marschalek, R.; Henderson, M.; et al. Effective targeting of the P53-MDM2 axis in preclinical models of infant MLL-rearranged acute lymphoblastic leukemia. Clin. Cancer Res. 2015, 21, 1395–1405. [Google Scholar] [CrossRef]
- Shimazu, K.; Tada, Y.; Morinaga, T.; Shingyoji, M.; Sekine, I.; Shimada, H.; Hiroshima, K.; Namiki, T.; Tatsumi, K.; Tagawa, M. Metformin produces growth inhibitory effects in combination with nutlin-3a on malignant mesothelioma through a cross-talk between mTOR and p53 pathways. BMC Cancer 2017, 17, 309. [Google Scholar] [CrossRef]
- Guo, Y.; Li, Y.; Wang, F.-F.; Xiang, B.; Huang, X.-O.; Ma, H.-B.; Gong, Y.-P. The combination of Nutlin-3 and Tanshinone IIA promotes synergistic cytotoxicity in acute leukemic cells expressing wild-type p53 by co-regulating MDM2-P53 and the AKT/mTOR pathway. Int. J. Biochem. Cell Biol. 2019, 106, 8–20. [Google Scholar] [CrossRef]
- Nguyen, D.; Liao, W.; Zeng, S.X.; Lu, H. Reviving the guardian of the genome: Small molecule activators of p53. Pharmacol. Ther. 2017, 178, 92–108. [Google Scholar] [CrossRef]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Estrada-Ortiz, N.; Neochoritis, C.G.; Dömling, A. How to Design a Successful p53-MDM2/X Interaction Inhibitor: A Thorough Overview Based on Crystal Structures. ChemMedChem 2016, 11, 757–772. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef]
- Ding, Q.; Zhang, Z.; Liu, J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA. 2008, 105, 3933–3938. [Google Scholar] [CrossRef]
- Canner, J.A.; Sobo, M.; Ball, S.; Hutzen, B.; DeAngelis, S.; Willis, W.; Studebaker, A.W.; Ding, K.; Wang, S.; Yang, D.; et al. MI-63: A novel small-molecule inhibitor targets MDM2 and induces apoptosis in embryonal and alveolar rhabdomyosarcoma cells with wild-type p53. Br. J. Cancer 2009, 101, 774–781. [Google Scholar] [CrossRef]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef]
- Aguilar, A.; Lu, J.; Liu, L.; Du, D.; Bernard, D.; McEachern, D.; Przybranowski, S.; Li, X.; Luo, R.; Wen, B.; et al. Discovery of 4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-ethyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)bicyclo[2.2.2]octane-1-carboxylic Acid (AA-115/APG-115): A Potent and Orally Active Murine Double Minut. J. Med. Chem. 2017, 60, 2819–2839. [Google Scholar] [CrossRef]
- Arnhold, V.; Schmelz, K.; Proba, J.; Winkler, A.; Wünschel, J.; Toedling, J.; Deubzer, H.E.; Künkele, A.; Eggert, A.; Schulte, J.H.; et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 2018, 9, 2304–2319. [Google Scholar] [CrossRef]
- Furet, P.; Chène, P.; De Pover, A.; Valat, T.S.; Lisztwan, J.H.; Kallen, J.; Masuya, K. The central valine concept provides an entry in a new class of non peptide inhibitors of the p53-MDM2 interaction. Bioorg. Med. Chem. Lett. 2012, 22, 3498–3502. [Google Scholar] [CrossRef]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef]
- Furet, P.; Masuya, K.; Kallen, J.; Stachyra-Valat, T.; Ruetz, S.; Guagnano, V.; Holzer, P.; Mah, R.; Stutz, S.; Vaupel, A.; et al. Discovery of a novel class of highly potent inhibitors of the p53-MDM2 interaction by structure-based design starting from a conformational argument. Bioorg. Med. Chem. Lett. 2016, 26, 4837–4841. [Google Scholar] [CrossRef]
- Swift, L.H.; Golsteyn, R.M. Genotoxic anti-cancer agents and their relationship to DNA damage, mitosis, and checkpoint adaptation in proliferating cancer cells. Int. J. Mol. Sci. 2014, 15, 3403–3431. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Astolfi, L.; Ghiselli, S.; Guaran, V.; Chicca, M.; Simoni, E.; Olivetto, E.; Lelli, G.; Martini, A. Correlation of adverse effects of cisplatin administration in patients affected by solid tumours: A retrospective evaluation. Oncol. Rep. 2013, 29, 1285–1292. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- Froelich-Ammon, S.J.; Osheroff, N. Topoisomerase poisons: Harnessing the dark side of enzyme mechanism. J. Biol. Chem. 1995, 270, 21429–21432. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef]
- Gianfaldoni, S.; Gianfaldoni, R.; Wollina, U.; Lotti, J.; Tchernev, G.; Lotti, T. An Overview on Radiotherapy: From Its History to Its Current Applications in Dermatology. Open Access Maced. J. Med. Sci. 2017, 5, 521–525. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Berkey, F.J. Managing the adverse effects of radiation therapy. Am. Fam. Physician 2010, 82, 381–388, 394. [Google Scholar]
- Perry, M.E.; Piette, J.; Zawadzki, J.A.; Harvey, D.; Levine, A.J. The mdm-2 gene is induced in response to UV light in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 2006, 90, 11623–11627. [Google Scholar] [CrossRef]
- Cao, C.; Shinohara, E.T.; Subhawong, T.K.; Geng, L.; Kim, K.W.; Albert, J.M.; Hallahan, D.E.; Lu, B. Radiosensitization of lung cancer by nutlin, an inhibitor of murine double minute 2. Mol. Cancer Ther. 2006, 5, 411–417. [Google Scholar] [CrossRef]
- Yi, H.; Yan, X.; Luo, Q.; Yuan, L.; Li, B.; Pan, W.; Zhang, L.; Chen, H.; Wang, J.; Zhang, Y.; et al. A novel small molecule inhibitor of MDM2-p53 (APG-115) enhances radiosensitivity of gastric adenocarcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 97. [Google Scholar] [CrossRef]
- Tangutoori, S.; Baldwin, P.; Sridhar, S. PARP inhibitors: A new era of targeted therapy. Maturitas 2015, 81, 5–9. [Google Scholar] [CrossRef]
- Shirley, M. Rucaparib: A Review in Ovarian Cancer. Target Oncol. 2019, 14, 237–246. [Google Scholar] [CrossRef]
- Pecháčková, S.; Burdová, K.; Macurek, L. WIP1 phosphatase as pharmacological target in cancer therapy. J. Mol. Med. 2017, 95, 589–599. [Google Scholar] [CrossRef]
- Richter, M.; Dayaram, T.; Gilmartin, A.G.; Ganji, G.; Pemmasani, S.K.; Van Der Key, H.; Shohet, J.M.; Donehower, L.A.; Kumar, R. WIP1 phosphatase as a potential therapeutic target in neuroblastoma. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Cory, S.; Huang, D.C.S.; Adams, J.M. The Bcl-2 family: Roles in cell survival and oncogenesis. Oncogene 2003, 22, 8590–8607. [Google Scholar] [CrossRef]
- Xu, X.; Lai, Y.; Hua, Z.-C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 1–8. [Google Scholar] [CrossRef]
- Niu, X.; Wang, G.; Wang, Y.; Caldwell, J.T.; Edwards, H.; Xie, C.; Taub, J.W.; Li, C.; Lin, H.; Ge, Y. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia 2014, 28, 1557–1560. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Haling, J.R.; Chen, H.; Song, K.; Price, S.; Heald, R.; Hewitt, J.F.M.; Zak, M.; Peck, A.; Orr, C.; et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS-versus BRAF-driven cancers. Nature 2013, 501, 232–236. [Google Scholar] [CrossRef]
- van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S.; et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Verstovsek, S.; Golemovic, M.; Kantarjian, H.; Manshouri, T.; Estrov, Z.; Manley, P.; Sun, T.; Arlinghaus, R.B.; Alland, L.; Dugan, M.; et al. AMN107, a novel aminopyrimidine inhibitor of p190 Bcr-Abl activation and of in vitro proliferation of Philadelphia-positive acute lymphoblastic leukemia cells. Cancer 2005, 104, 1230–1236. [Google Scholar] [CrossRef]
- Golemovic, M.; Giles, F.; Cortes, J.; Manshouri, T.; Manley, P.W.; Dugan, M.; Alland, L.; Griffin, J.D.; Arlinghaus, R.B.; Sun, T.; et al. Cancer Therapy: Preclinical AMN107, a Novel Aminopyrimidine Inhibitor of Bcr-Abl, Has In vitro Activity against Imatinib-Resistant Chronic Myeloid Leukemia. Clin. Cancer Res. 2005, 11, 4941–4947. [Google Scholar] [CrossRef]
- Gu, Z.; Wang, X.; Qi, R.; Wei, L.; Huo, Y.; Ma, Y.; Shi, L.; Chang, Y.; Li, G.; Zhou, L. Oridonin induces apoptosis in uveal melanoma cells by upregulation of Bim and downregulation of Fatty Acid Synthase. Biochem. Biophys. Res. Commun. 2015, 457, 187–193. [Google Scholar] [CrossRef]
- Weng, H.; Huang, H.; Dong, B.; Zhao, P.; Zhou, H.; Qu, L. Inhibition of miR-17 and miR-20a by oridonin triggers apoptosis and reverses chemoresistance by derepressing BIM-S. Cancer Res. 2014, 74, 4409–4419. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activationby Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef]
- Riley, M.F.; Lozano, G. The Many Faces of MDM2 Binding Partners. Genes Cancer 2012, 3, 226–239. [Google Scholar] [CrossRef]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Kimberley, F.C.; Screaton, G.R. Following a TRAIL: Update on a ligand and its five receptors. Cell Res. 2004, 14, 359–372. [Google Scholar] [CrossRef]
- De Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef]
- Merino, D.; Lalaoui, N.; Morizot, A.; Schneider, P.; Solary, E.; Micheau, O. Differential Inhibition of TRAIL-Mediated DR5-DISC Formation by Decoy Receptors 1 and 2. Mol. Cell. Biol. 2006, 26, 7046–7055. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Wilson, N.S.; Dixit, V.; Ashkenazi, A. Death receptor signal transducers: Nodes of coordination in immune signaling networks. Nat. Immunol. 2009, 10, 348–355. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Treon, S.P.; Mitsiades, N.; Shima, Y.; Richardson, P.; Schlossman, R.; Hideshima, T.; Anderson, K.C. TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug resistance in multiple myeloma: Therapeutic applications. Blood 2001, 98, 795–804. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Holland, P.; Eckhardt, S.G. Ligand-Based Targeting of Apoptosis in Cancer: The Potential of Recombinant Human Apoptosis Ligand 2/Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand (rhApo2L/TRAIL). J. Clin. Oncol. 2008, 26, 3621–3630. [Google Scholar] [CrossRef]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef]
- Pan, Y.; Xu, R.; Peach, M.; Huang, C.P.; Branstetter, D.; Novotny, W.; Herbst, R.S.; Eckhardt, S.G.; Holland, P.M. Evaluation of pharmacodynamic biomarkers in a Phase 1a trial of dulanermin (rhApo2L/TRAIL) in patients with advanced tumours. Br. J. Cancer 2011, 105, 1830–1838. [Google Scholar] [CrossRef]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef]
- Kay, B.P.; Hsu, C.P.; Lu, J.F.; Sun, Y.N.; Bai, S.; Xin, Y.; D’Argenio, D.Z. Intracellular-signaling tumor-regression modeling of the pro-apoptotic receptor agonists dulanermin and conatumumab. J. Pharmacokinet. Pharmacodyn. 2012, 39, 577–590. [Google Scholar] [CrossRef][Green Version]
- van der Sloot, A.M.; Tur, V.; Szegezdi, E.; Mullally, M.M.; Cool, R.H.; Samali, A.; Serrano, L.; Quax, W.J. Designed tumor necrosis factor-related apoptosis-inducing ligand variants initiating apoptosis exclusively via the DR5 receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 8634–8639. [Google Scholar] [CrossRef]
- Bozic, I. Dynamics of Targeted Cancer Therapy. Trends Mol. Med. 2012, 18, 311–316. [Google Scholar] [CrossRef][Green Version]
- Yan, L.; Rosen, N.; Arteaga, C. Targeted cancer therapies. Chin. J. Cancer 2011, 30, 1–4. [Google Scholar] [CrossRef]
- Viswanadha, V.P. An overview of targeted cancer therapy. BioMedicine 2015, 5, 1–6. [Google Scholar]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14, 57–66. [Google Scholar] [CrossRef]
- Krause, D.S.; Van Etten, R.A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef]
- Sangwan, V.; Park, M. Receptor tyrosine kinases: Role in cancer progression. Curr. Oncol. 2006, 13, 191–193. [Google Scholar]
- Manash, P.; Anup, M. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar]
- Bose, P.; Gandhi, V.V.; Keating, M.J. Pharmacokinetic and pharmacodynamic evaluation of ibrutinib for the treatment of chronic lymphocytic leukemia: rationale for lower doses. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1381–1392. [Google Scholar] [CrossRef]
- Allocati, N.; Di Ilio, C.; De Laurenzi, V. P63/p73 in the control of cell cycle and cell death. Exp. Cell Res. 2012, 318, 1285–1290. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef]
- Yu, Z.; Ye, S.; Hu, G.; Lv, M.; Tu, Z.; Zhou, K.; Li, Q. The RAF-MEK-ERK pathway: Targeting ERK to overcome obstacles to effective cancer therapy. Future Med. Chem. 2015, 7, 269–289. [Google Scholar] [CrossRef]
- Muta, Y.; Matsuda, M.; Imajo, M. Divergent Dynamics and Functions of ERK MAP Kinase Signaling in Development, Homeostasis and Cancer: Lessons from Fluorescent Bioimaging. Cancers 2019, 11, 513. [Google Scholar] [CrossRef]
- Lee, J.T.; Herlyn, M. MEK’ing the most of p53 reactivation therapy in melanoma. J. Invest. Dermatol. 2012, 132, 263–265. [Google Scholar] [CrossRef]
- Davies, B.R.; Logie, A.; McKay, J.S.; Martin, P.; Steele, S.; Jenkins, R.; Cockerill, M.; Cartlidge, S.; Smith, P.D. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: Mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical. Mol. Cancer Ther. 2007, 6, 2209–2219. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 1–11. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef]
- Khan, K.H.; Yap, T.A.; Yan, L.; Cunningham, D. Targeting the PI3K-AKT-mTOR singnaling network in cancer. Chin. J. Cancer 2013, 32, 253–265. [Google Scholar] [CrossRef]
- Richardson, P. Perifosine, an oral, anti-cancer agent and inhibitor of the Akt pathway: Mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 623–633. [Google Scholar] [CrossRef]
- Sánchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; De Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorganic Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar]
- Koivunen, J.; Aaltonen, V.; Peltonen, J. Protein kinase C (PKC) family in cancer progression. Cancer Lett. 2006, 235, 1–10. [Google Scholar] [CrossRef]
- Cooke, M.; Magimaidas, A.; Casado-Medrano, V.; Kazanietz, M.G. Protein kinase C in cancer: The top five unanswered questions. Mol. Carcinog. 2017, 56, 1531–1542. [Google Scholar] [CrossRef]
- Isakov, N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52. [Google Scholar] [CrossRef]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef]
- Scott, A.M.; Allison, J.P.; Wolchok, J.D. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012, 12, 1–8. [Google Scholar]
- Chiavenna, S.M.; Jaworski, J.P.; Vendrell, A. State of the art in anti-cancer mAbs. J. Biomed. Sci. 2017, 24, 1–12. [Google Scholar] [CrossRef]
- Nicodemus, C.F. Antibody-based immunotherapy of solid cancers: Progress and possibilities. Immunotherapy 2015, 7, 923–939. [Google Scholar] [CrossRef]
- Cuesta-Mateos, C.; Alcaraz-Serna, A.; Somovilla-Crespo, B.; Muñoz-Calleja, C. Monoclonal antibody therapies for hematological malignancies: Not just lineage-specific targets. Front. Immunol. 2018, 8, 1936. [Google Scholar] [CrossRef]
- New Clinical Trials of Monoclonal Antibodies Have Grown 115% in the Last Ten Years. Available online: https://www.globaldata.com/new-clinical-trials-monoclonal-antibodies-grown-115-last-ten-years/ (accessed on 18 July 2019).
- Konstantinidou, M.; Zarganes-Tzitzikas, T.; Magiera-Mularz, K.; Holak, T.A.; Dömling, A. Immune Checkpoint PD-1/PD-L1: Is There Life Beyond Antibodies? Angew. Chem. Int. Ed. Engl. 2018, 57, 4840–4848. [Google Scholar] [CrossRef]
- Freeman, C.L.; Sehn, L.H. A tale of two antibodies: Obinutuzumab versus rituximab. Br. J. Haematol. 2018, 182, 29–45. [Google Scholar] [CrossRef]
- Gagez, A.-L.; Cartron, G. Obinutuzumab: A new class of anti-CD20 monoclonal antibody. Curr. Opin. Oncol. 2014, 26, 484–491. [Google Scholar] [CrossRef]
- Owen, C.J.; Stewart, D.A. Obinutuzumab for the treatment of patients with previously untreated chronic lymphocytic leukemia: Overview and perspective. Ther. Adv. Hematol. 2015, 6, 161–170. [Google Scholar] [CrossRef]
- Pirich, T.; Zwickl-Traxler, E.; Pecherstorfer, M.; Singer, J. Tolerability of obinutuzumab therapy in patients with rituximab-relapsed/refractory B-cell malignancies—A retrospective single center cohort study. Oncotarget 2018, 9, 29944–29956. [Google Scholar] [CrossRef][Green Version]
- Williams, R. Discontinued drugs in 2012: Oncology drugs. Expert Opin. Investig. Drugs 2013, 22, 1627–1644. [Google Scholar] [CrossRef]
- Papandreou, C.N. The Proteasome as a Target for Cancer Treatment. Am. J. Cancer 2005, 4, 359–372. [Google Scholar] [CrossRef][Green Version]
- Richardson, P.G.; Mitsiades, C.; Hideshima, T.; Anderson, K.C. Proteasome inhibition in the treatment of cancer. Cell Cycle 2005, 4, 290–296. [Google Scholar] [CrossRef]
- Orlowski, R.Z.; Kuhn, D.J. Proteasome inhibitors in cancer therapy: Lessons from the first decade. Clin. Cancer Res. 2008, 14, 1649–1657. [Google Scholar] [CrossRef]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Manal, M.; Chandrasekar, M.J.N.; Gomathi Priya, J.; Nanjan, M.J. Inhibitors of histone deacetylase as antitumor agents: A critical review. Bioorg. Chem. 2016, 67, 18–42. [Google Scholar] [CrossRef]
- Brodie, S.; Brandes, J. Could valproic acid be an effective anticancer agent? The evidence so far. Expert Rev. Anticancer Ther. 2014, 14, 1097–1100. [Google Scholar] [CrossRef]
- Kleeff, J.; Kornmann, M.; Sawhney, H.; Korc, M. Actinomycin D induces apoptosis and inhibits growth of pancreatic cancer cells. Int. J. Cancer 2000, 86, 399–407. [Google Scholar] [CrossRef]
- Liu, X.F.; Xiang, L.; Zhou, Q.; Carralot, J.-P.; Prunotto, M.; Niederfellner, G.; Pastan, I. Actinomycin D enhances killing of cancer cells by immunotoxin RG7787 through activation of the extrinsic pathway of apoptosis. Proc. Natl. Acad. Sci. USA 2016, 113, 10666–10671. [Google Scholar] [CrossRef]
- Bosschieter, J.; Nieuwenhuijzen, J.A.; van Ginkel, T.; Vis, A.N.; Witte, B.; Newling, D.; Beckers, G.M.A.; van Moorselaar, R.J.A. Value of an Immediate Intravesical Instillation of Mitomycin C in Patients with Non-muscle-invasive Bladder Cancer: A Prospective Multicentre Randomised Study in 2243 patients. Eur. Urol. 2018, 73, 226–232. [Google Scholar] [CrossRef]
- Lu, D.; Wang, Y.; Li, C.; Wei, G.; Chen, R.; Dong, D.; Yao, M. Actinomycin D inhibits cell proliferations and promotes apoptosis in osteosarcoma cells. Int. J. Clin. Exp. Med. 2015, 8, 1904–1911. [Google Scholar]
- Loh, S.N. The missing Zinc: P53 misfolding and cancer. Metallomics 2010, 2, 442–449. [Google Scholar] [CrossRef]
- Morita, A.; Ariyasu, S.; Ohya, S.; Takahashi, I.; Wang, B.; Tanaka, K.; Uchida, T.; Okazaki, H.; Hanaya, K.; Enomoto, A.; et al. Evaluation of zinc (II) chelators for inhibiting p53-mediated apoptosis. Oncotarget 2013, 4, 2439–2450. [Google Scholar] [CrossRef]
- Kogan, S.; Carpizo, D.R. Zinc Metallochaperones as Mutant p53 Reactivators: A New Paradigm in Cancer Therapeutics. Cancers 2018, 10, 166. [Google Scholar] [CrossRef]
- Adlard, P.A.; Bush, A.I. Metal chaperones: A holistic approach to the treatment of Alzheimer’s disease. Front. psychiatry 2012, 3, 15. [Google Scholar] [CrossRef]
- Lowe, S.W.; Ruley, H.E.; Jacks, T.; Housman, D.E. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993, 74, 957–967. [Google Scholar] [CrossRef]
- Whitley, D.; Goldberg, S.P.; Jordan, W.D. Heat shock proteins: A review of the molecular chaperones. J. Vasc. Surg. 1999, 29, 748–751. [Google Scholar] [CrossRef]
- Schneider, C.; Sepp-Lorenzino, L.; Nimmesgern, E.; Ouerfelli, O.; Danishefsky, S.; Rosen, N.; Hartl, F.U. Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc. Natl. Acad. Sci. USA 1996, 93, 14536–14541. [Google Scholar] [CrossRef]
- Flandrin, P.; Guyotat, D.; Duval, A.; Cornillon, J.; Tavernier, E.; Nadal, N.; Campos, L. Significance of heat-shock protein (HSP) 90 expression in acute myeloid leukemia cells. Cell Stress Chaperones 2008, 13, 357–364. [Google Scholar] [CrossRef]
- Wiedmann, R.M.; von Schwarzenberg, K.; Palamidessi, A.; Schreiner, L.; Kubisch, R.; Liebl, J.; Schempp, C.; Trauner, D.; Vereb, G.; Zahler, S.; et al. The V-ATPase-inhibitor archazolid abrogates tumor metastasis via inhibition of endocytic activation of the Rho-GTPase Rac1. Cancer Res. 2012, 72, 5976–5987. [Google Scholar] [CrossRef]
- Schempp, C.M.; von Schwarzenberg, K.; Schreiner, L.; Kubisch, R.; Muller, R.; Wagner, E.; Vollmar, A.M. V-ATPase Inhibition Regulates Anoikis Resistance and Metastasis of Cancer Cells. Mol. Cancer Ther. 2014, 13, 926–937. [Google Scholar] [CrossRef]
- Bartel, K.; Winzi, M.; Ulrich, M.; Koeberle, A.; Menche, D.; Werz, O.; Müller, R.; Guck, J.; Vollmar, A.M.; von Schwarzenberg, K. V-ATPase inhibition increases cancer cell stiffness and blocks membrane related Ras signaling—A new option for HCC therapy. Oncotarget 2017, 8, 9476–9487. [Google Scholar] [CrossRef]
- Kasper, E.; Angot, E.; Colasse, E.; Nicol, L.; Sabourin, J.C.; Adriouch, S.; Lacoume, Y.; Charbonnier, C.; Raad, S.; Frebourg, T.; et al. Contribution of genotoxic anticancer treatments to the development of multiple primary tumours in the context of germline TP53 mutations. Eur. J. Cancer 2018, 101, 254–262. [Google Scholar] [CrossRef]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy (review). Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.M.; Pellegrini, F.; Nicolucci, A. Metformin Therapy and Risk of Cancer in Patients with Type 2 Diabetes: Systematic Review. PLoS ONE 2013, 8, 1–12. [Google Scholar] [CrossRef]
- Lane, D.P.; Cheok, C.F.; Lain, S. P53-Based Cancer Therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, 1–24. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode of Action | Combination | In Vitro Studies | In Vivo Studies | Clinical Trials | References | ||
|---|---|---|---|---|---|---|---|
| Therapeutic Agent | HDM2 Antagonist | ||||||
| DNA Damage | 5-azacitadine | DS-3032b | - | - | AML or MDS, Phase 1 | NCT02319369 | |
| 5-fluorouracil | CGM097 | neuroendocrine tumor cell lines | - | - | [32] | ||
| acadesine | nutlin-3a | CLL | - | - | [33] | ||
| busulfan | idasanutlin | neuroblastoma | - | - | [34] | ||
| carboplatin | AMG-232 | different tumor cell lines | osteosarcoma, colorectal carcinoma, melanoma, NSCLC xenograft mouse models | - | [35] | ||
| APG-115 | - | - | salivary gland carcinoma, Phase 1/2 | NCT03781986 | |||
| nutlin-3a | breast cancer | breast cancer xenograft mouse models | - | [36] | |||
| cisplatin | AMG-232 | different tumor cell lines | osteosarcoma, colorectal carcinoma, melanoma, NSCLC xenograft mouse models | - | [35] | ||
| idasanutlin | ovarian cancer, neuroblastoma | - | - | [34,37] | |||
| MI-319 | pancreatic cancer | - | - | [38] | |||
| nutlin-3 | nasopharyngeal carcinoma, adenocarcinoma, ovarian cancer | - | - | [37,39,40,41] | |||
| nutlin-3a | liposarcoma, RMS, neuroblastoma, osteosarcoma | - | - | [42,43] | |||
| chlorambucil | nutlin-3a | CLL | - | - | [33] | ||
| CPT-11 | AMG-232 | different tumor cell lines | osteosarcoma, colorectal carcinoma, melanoma, NSCLC xenograft mouse models | - | [35] | ||
| cytarabine | ALRN-6924 | - | - | AML and AMS, Phase 1 | NCT02909972 | ||
| CGM097 | AML | - | - | [44] | |||
| HDM-201 | AML | - | AML, Phase 1/2 | [44] NCT03760445 | |||
| idasanutlin | - | - | AML, Phase 1/2, Phase 3 | NCT03850535 NCT02545283 | |||
| RG7112 | - | - | AML, Phase 1 | NCT01635296 | |||
| daunorubicin | HDM-201 | - | - | AML, Phase 1/2 | NCT03760445 | ||
| idasanutlin | - | - | AML, Phase 1/2 | NCT03850535 | |||
| DNA Damage | decitabine | AMG-232 | - | - | AML, Phase 1 | NCT03041688 | |
| doxorubicin | AMG-232 | different tumor cell lines | osteosarcoma, colorectal carcinoma, melanoma, NSCLC xenograft mouse models | - | [35] | ||
| idasanutlin | neuroblastoma | - | - | [34] | |||
| nutlin-3a | DLBCL, CLL, PDAC, liposarcoma, RMS and osteosarcoma | DLBCL xenograft mouse models | - | [33,42,45,46] | |||
| etoposide | MI-219 | lung cancer | - | - | [47] | ||
| nutlin-3 | ovarian cancer | - | - | [41] | |||
| nutlin-3a | PDAC, neuroblastoma | - | - | [43,46] | |||
| fludarabine | nutlin-3a | CLL | - | - | [33,48] | ||
| gemcitabine | MI-63 | MCL | - | - | [49] | ||
| nutlin-3a | PDAC | - | - | [46] | |||
| idarubicin | CGM097 | AML | - | - | [44] | ||
| HDM-201 | |||||||
| ionizing radiation (IR) | AMG-232 | - | adenoid cystic carcinoma xenograft mouse models | glioblastoma, Phase 1, soft tissue sarcoma, Phase 1 | [50] NCT03107780 NCT03217266 | ||
| RG7388 | - | RMS xenograft mouse models | - | [51] | |||
| nutlin-3 | LSCC, glioblastoma | - | - | [52,53] | |||
| oxaliplatin | MI-219 | PDAC, colon and breast cancer | - | - | [54] | ||
| temozolomide | CGM097 | neuroendocrine tumor cell lines | - | - | [32] | ||
| idasanutlin | neuroblastoma | - | - | [34] | |||
| RG7775 | neuroblastoma | neuroblastoma mouse xenograft models | - | [55] | |||
| topotecan | idasanutlin | neuroblastoma | - | - | [34] | ||
| trabectedin | RG7112 | fibrosarcoma and liposarcoma | - | - | [56] | ||
| DNA Damage Response Sustaining | GSK2830371 | ATSP-7041 | Ewing’s sarcoma | - | - | [57] | |
| HDM-201 | human melanoma | - | - | [15] | |||
| idasanutlin | |||||||
| nutlin-3 | |||||||
| nutlin-3a | osteosarcoma, breast, colon adenocarcinoma | - | - | [58] | |||
| rucaparib | idasanutlin | ovarian cancer | - | - | [59] | ||
| nutlin-3 | |||||||
| Apoptosis Induction | BCL-2 Inhibitors | ABT-263 | SAR405838 | ALL and AML | ALL and AML xenograft mouse models | - | [10] |
| ABT-737 | MI-63 | multiple myeloma | multiple myeloma xenograft mouse models | - | [60,61,62] | ||
| nutlin-3 | AML, CML | - | - | [60,61] | |||
| BCL-2 Dowregulators | oridonin | nutlin-3 | osteosarcoma | - | - | [63] | |
| venetoclax | idasanutlin | AML, neuroblastoma | AML, neuroblastoma xenograft mouse models | AML patients, Phase 1b/2 | [64,65] NCT02670044 | ||
| venetoclax + obinutuzumab or rituximab | idasanutlin | - | - | follicular lymphoma and DLBCL | NCT03135262 | ||
| SMAC Mimetics | 1396-11 | nutlin-3a | AML | - | - | [66] | |
| ABT-10 | |||||||
| SM-164 | MI-219 | lung cancer | - | - | [47] | ||
| TRAIL Agonists | D269H/E195R | nutlin-3 | NSCLC, colon, ovarian cancer | - | - | [67] | |
| rhTRAIL | nutlin-3 | NSCLC, colon, ovarian cancer | - | - | [67] | ||
| nutlin-3a | MPM | - | - | [68] | |||
| RG7112 | - | MPM xenograft mouse models | - | [68] | |||
| Pro-survival Signaling Pathways Targeting | Tyrosine Kinase Inhibitors | alpelisib | CGM097 | AML | - | - | [44] |
| HDM-201 | |||||||
| buparlisib | CGM097 | AML | - | - | [44] | ||
| HDM-201 | |||||||
| ceritinib | CGM097 | neuroblastoma | neuroblastoma xenograft mouse models | - | [69] | ||
| dasatinib | nutlin-3 | B-CLL | - | - | [70] | ||
| nutlin-3a | ALL | - | - | [71] | |||
| gilterinib | CGM097 | AML | - | - | [44] | ||
| HDM-201 | |||||||
| ibrutinib | nutlin-3 | CLL | CLL xenograft mouse models | - | [72] | ||
| imatinib | nutlin-3a | ALL | - | - | [71] | ||
| midostaurin | CGM097 | AML | AML xenograft mouse models | - | [44,73] | ||
| HDM-201 | AML | - | AML, Phase 1/2 | [44] NCT03760445 | |||
| nilotinib | nutlin-3 | AML, CML | - | - | [60,61] | ||
| nutlin-3a | ALL | - | - | [71] | |||
| pictilisib | CGM097 | AML | - | - | [44] | ||
| HDM-201 | |||||||
| Pro-Survival Signaling Pathways Targeting | Tyrosine Kinase Inhibitors | quizartinib | CGM097 | AML | AML xenograft mouse models | - | [73] |
| DS-3032b | - | - | AML, Phase 1 | NCT03552029 | |||
| ruxolitinib | KRT232 | - | - | PV, Phase 2a/2b | NCT03669965 | ||
| sorafenib | nutlin-3 | AML, RCC | - | - | [74,75] | ||
| Ras/Raf/MEK/MAPK Inhibitors | AZD6244 | CGM097 | AML | - | - | [76] | |
| nutlin-3a | AML | - | - | [77] | |||
| dabrafenib | AMG-232 | different human cancer cell lines | colorectal cancer xenograft mouse models | melanoma, Phase 1 | NCT02110355 [78,79] | ||
| LGX818 | CGM097 | melanoma | melanoma mouse models | - | [80] | ||
| PD0325901 | AMG-232 | different human cancer cell lines | colorectal cancer xenograft mouse models | - | [79] | ||
| pimasertib | SAR405838 | - | - | different tumor types, Phase 1 | NCT01985191 [81] | ||
| trametinib | AMG-232 | different human cancer cell lines | colorectal cancer xenograft mouse models | AML, Phase 1b melanoma, Phase 1 | [78,79] NCT02016729 NCT02110355 | ||
| HDM-201 | cutaneous melanoma | - | colorectal carcinoma, Phase 1 | [82] NCT03714958 | |||
| idasanutlin | cutaneous melanoma | - | - | [82] | |||
| nutlin-3 | |||||||
| vemurafenib | AMG-232 | different human cancer cell lines | colorectal cancer xenograft mouse models | - | [79] | ||
| PI3K/Akt/mTOR Inhibitors | AMG511 | AMG-232 | different human cancer cell lines | colorectal cancer xenograft mouse models | - | [79] | |
| AZD8055 | AMG-232 | different human cancer cell lines | colorectal cancer xenograft mouse models | - | [79] | ||
| BEZ235 | AMG-232 | different human cancer cell lines | colorectal cancer xenograft mouse models | - | [79] | ||
| idasanutlin | liposarcoma | liposarcoma xenograft mouse models | - | [83] | |||
| everolimus | CGM097 | neuroendocrine tumor | - | - | [32] | ||
| GDC-0941 | AMG-232 | different human cancer cell lines | colorectal cancer xenograft mouse models | - | [79] | ||
| Ly294002 | nutlin-3 | ALL | - | - | [84] | ||
| MK-2206 | AMG-232 | different human cancer cell lines | colorectal cancer xenograft mouse models | - | [79] | ||
| PI103 | nutlin-3 | AML | - | - | [85] | ||
| perifosine | nutlin-3 | AML, B-CLL | - | - | [86] | ||
| Pro-Survival Signaling Pathways Targeting | CDK Inhibitors | abemaciclib | nutlin-3a | liposarcoma | - | - | [87] |
| palbociclib | idasanutlin | liposarcoma | liposarcoma xenograft mouse models | - | [88] | ||
| nutlin-3a | liposarcoma | - | - | [87] | |||
| ribociclib | HDM-201 | - | - | liposarcomas, Phase 1b/2 | NCT02343172 | ||
| nutlin-3a | liposarcoma | - | - | [87] | |||
| seliciclib | nutlin-3 | neuroblastoma | - | - | [89] | ||
| nutlin-3a | melanoma, breast, colon adenocarcinoma, liver carcinoma | - | - | [90] | |||
| PKC Inhibitors | LXS196 | HDM-201 | - | - | metastatic uveal melanoma, Phase 1 | NCT02601378 | |
| sotrastaurin | CGM097 | uveal melanoma | uveal melanoma xenograft mouse models | - | [91] | ||
| Therapeutic Antibodies | Anti-PD-1 and Anti PD-L1 | atezolizumab | idasanutlin | - | - | metastatic ER + breast cancer, Phase 1b/2 | NCT03566485 |
| pembrolizumab | APG-115 | - | - | unresectable or metastatic melanoma, advanced solid tumors, Phase 1b/2 | NCT03611868 | ||
| spartalizumab | HDM-201 | - | - | colorectal cancer, RCC Phase 1 | NCT02890069 | ||
| Anti-CD20 | obinutuzumab | nutlin-3 | CLL | - | - | [92] | |
| idasanutlin | MCL, DLBCL | MCL, DLBCL mouse xenograft models | FL, DLBCL, Phase 1b/2 | [93] NCT02624986 NCT03135262 | |||
| rituximab | nutlin-3 | CLL | - | - | [92] | ||
| idasanutlin | MCL, DLBCL | MCL, DLBCL mouse xenograft models | FL, DLBCL, Phase 1b/2 | [93] NCT02624986 NCT03135262 | |||
| Anti-DR5 | drozitumab | nutlin-3a | osteosarcoma, Ewing’s sarcoma | - | - | [94] | |
| Miscellaneous Anti-Cancer Agents | Proteasome Inhibitors | bortezomib | nutlin-3 | MCL, myeloma, breast, prostate, thyroid, colon cancer | - | - | [95,96,97] |
| carfilzomib | AMG-232 | - | - | multiple myeloma, Phase 1 | NCT03031730 | ||
| ixazomib citrate | idasanutlin | - | - | multiple myeloma, Phase 1/2 | NCT02633059 | ||
| MG-132 | nutlin-3 | schwannoma | schwannoma xenograft mouse models | - | [98] | ||
| HDACs Inhibitors | SAHA | idasanutlin | AML | - | - | [99] | |
| VPA | nutlin-3 | AML | AML xenograft mouse models | - | [100] | ||
| Antibiotics | actinomycin D | nutlin-3 | RMS | - | - | [101] | |
| different cancer cell lines | [102] | ||||||
| Zinc | zinc | MI-219 | colon and breast cancer | - | - | [38] | |
| ZMC1 | nutlin | ovarian, lung, nasopharyngeal cancer | ovarian xenograft mouse models | - | [103] | ||
| HSP Inhibitor | geldanamycin | nutlin-3 | AML | - | - | [104] | |
| ATP-ase Inhibitor | archazolid | nutlin-3a | liver, cervical, breast cancer, glioblastoma | glioblastoma xenograft models | - | [105] | |
| Mitotic Inhibitors | paclitaxel | ALRN-6924 | - | - | advanced, metastatic, or unresectable solid tumors, Phase 1 | NCT03725436 | |
| vincristine | nutlin-3 | RMS | - | - | [101] | ||
| RG7112 | leukemia cell lines | MLL-ALL xenograft mouse models | - | [106] | |||
| Others | metformin | nutlin-3a | malignant mesothelioma | - | - | [107] | |
| methotrexate | nutlin-3a | liposarcoma, RMS and osteosarcoma | - | - | [42] | ||
| tanshinone IIA | nutlin-3 | AML | - | - | [108] | ||
| P5091 | ATSP-7041 | Ewing’s sarcoma | - | - | [57] | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocik, J.; Machula, M.; Wisniewska, A.; Surmiak, E.; Holak, T.A.; Skalniak, L. Helping the Released Guardian: Drug Combinations for Supporting the Anticancer Activity of HDM2 (MDM2) Antagonists. Cancers 2019, 11, 1014. https://doi.org/10.3390/cancers11071014
Kocik J, Machula M, Wisniewska A, Surmiak E, Holak TA, Skalniak L. Helping the Released Guardian: Drug Combinations for Supporting the Anticancer Activity of HDM2 (MDM2) Antagonists. Cancers. 2019; 11(7):1014. https://doi.org/10.3390/cancers11071014
Chicago/Turabian StyleKocik, Justyna, Monika Machula, Aneta Wisniewska, Ewa Surmiak, Tad A. Holak, and Lukasz Skalniak. 2019. "Helping the Released Guardian: Drug Combinations for Supporting the Anticancer Activity of HDM2 (MDM2) Antagonists" Cancers 11, no. 7: 1014. https://doi.org/10.3390/cancers11071014
APA StyleKocik, J., Machula, M., Wisniewska, A., Surmiak, E., Holak, T. A., & Skalniak, L. (2019). Helping the Released Guardian: Drug Combinations for Supporting the Anticancer Activity of HDM2 (MDM2) Antagonists. Cancers, 11(7), 1014. https://doi.org/10.3390/cancers11071014