Systematic Analysis of Gene Expression in Lung Adenocarcinoma and Squamous Cell Carcinoma with a Case Study of FAM83A and FAM83B

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Lung Cancer Database
2.2. Meta-Analysis
2.3. Pathway Enrichment Analysis
2.4. Ethics Approval and Consent to Participate
2.5. Availability of Data and Material
3. Results
3.1. Quality Control and Reproducibility across Different Studies
3.1.1. Comparison between Pooled-Sample Analysis and Meta-Analysis
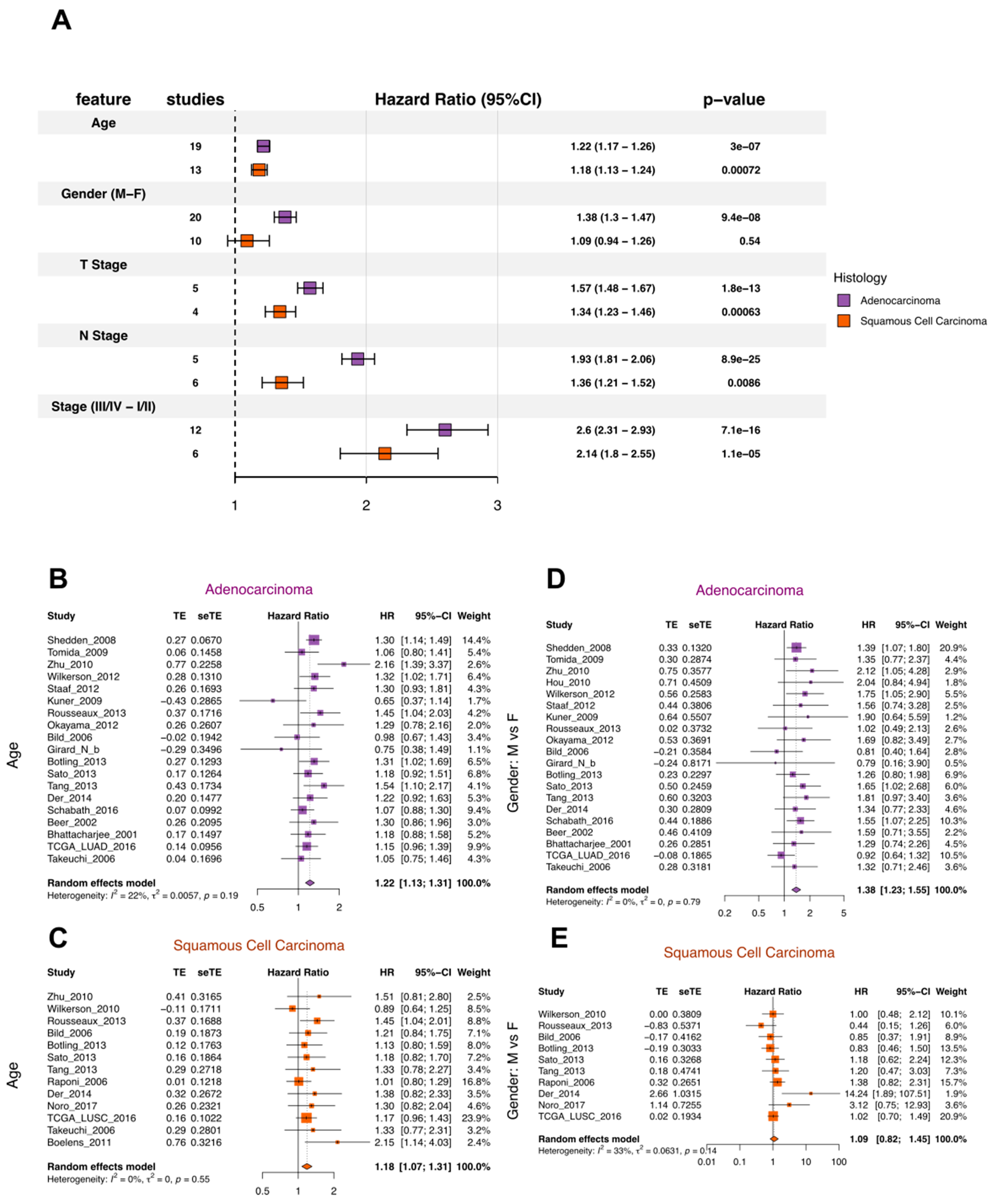
3.1.2. Quality Control for Clinical Variables and Survival Outcomes
3.1.3. Principal Component Analysis of the Transcriptomic Data Comparing Tumor and Normal Lung
3.1.4. Reproducibility in Tumor vs. Normal Expression Difference and Gene Expression-Survival Association
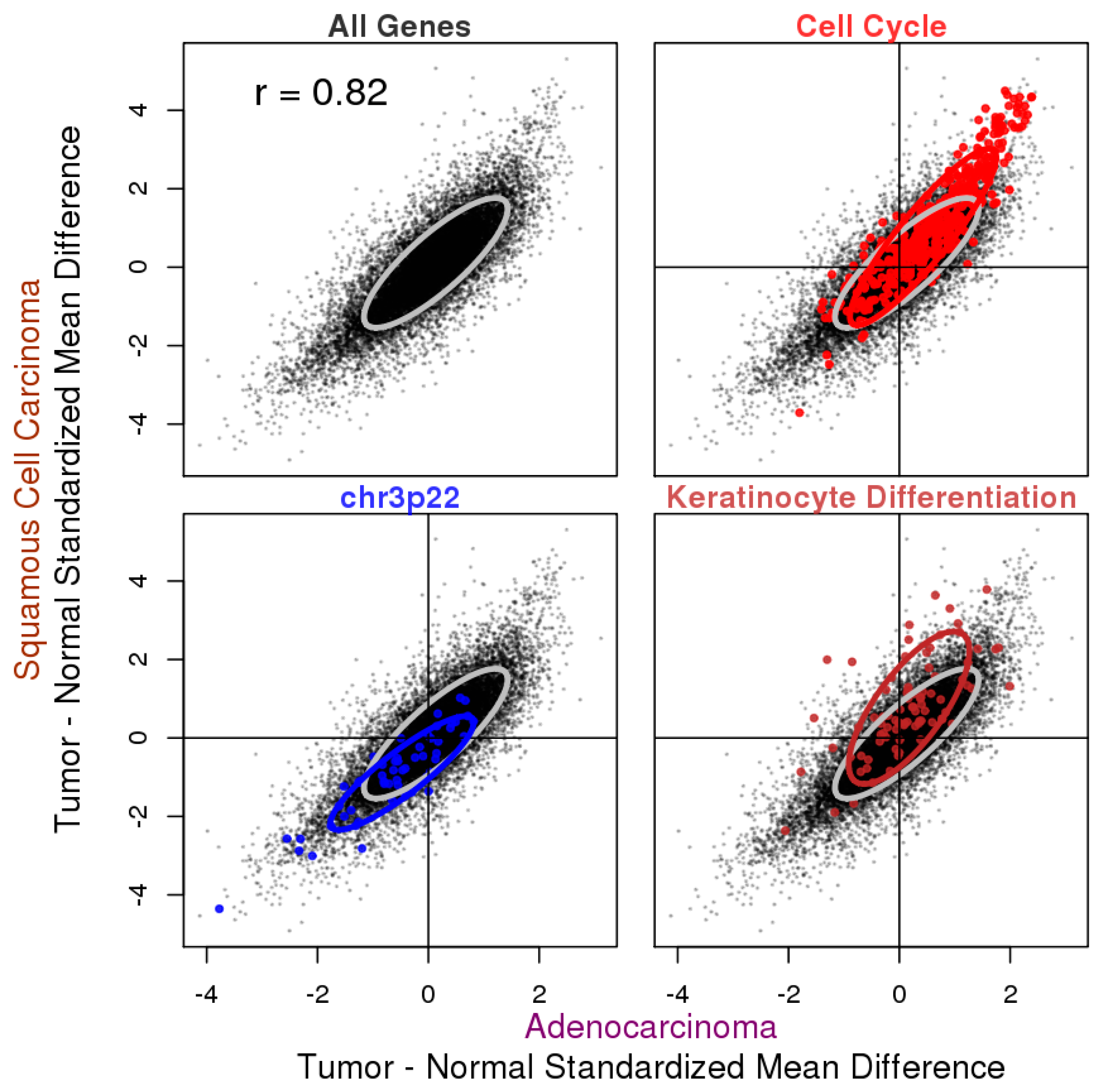
3.2. Tumor vs. Normal Expression Difference in ADC and SQCC
3.3. Gene Expression Association with Overall Survival in ADC and SQCC
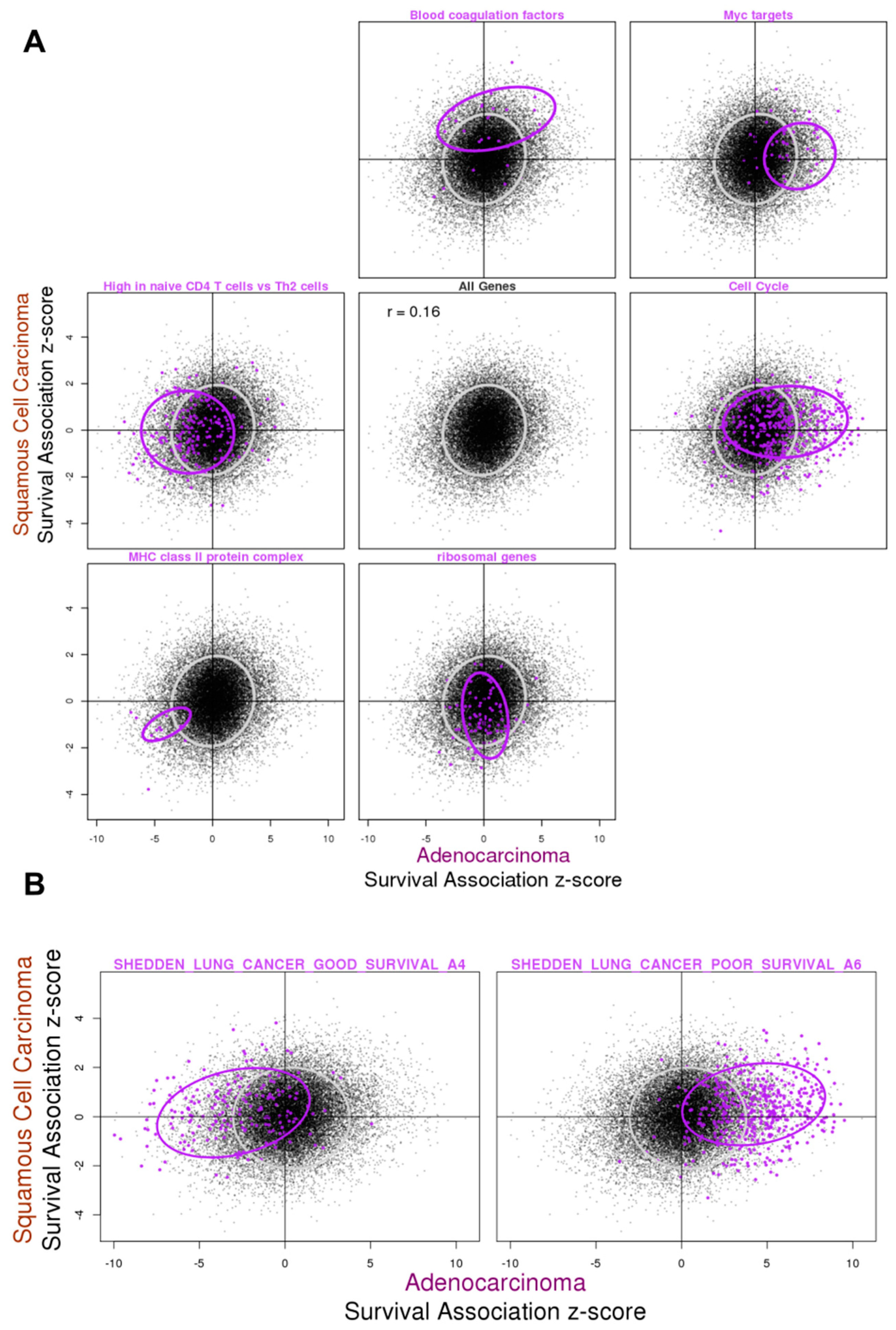
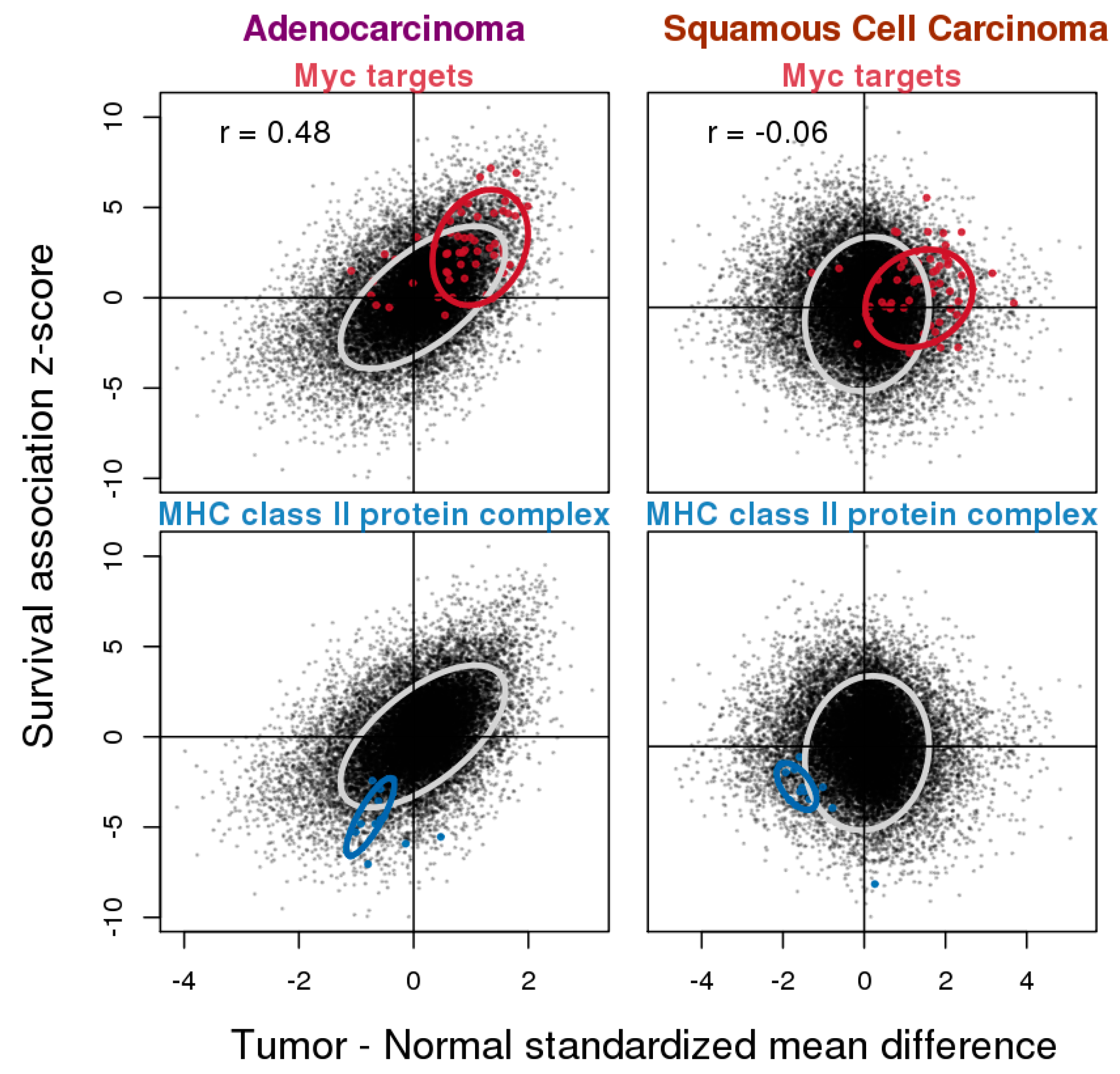
3.4. Relationship between Gene Expression Difference in Tumor vs. Normal and Association with Overall Survival
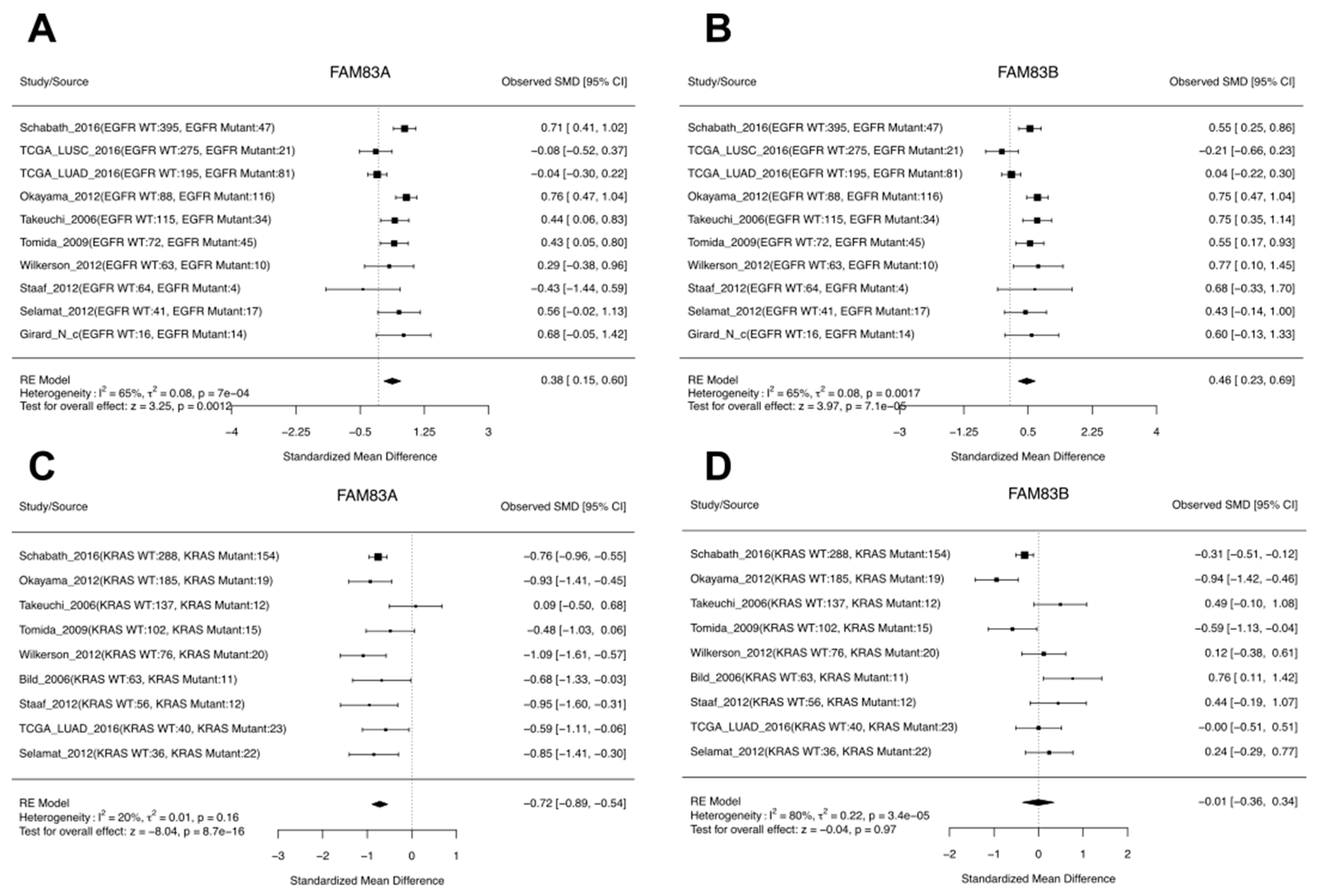
3.5. A Case Study of FAM83A and FAM83B Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cai, L.; Lin, S.; Girard, L.; Zhou, Y.; Yang, L.; Ci, B.; Zhou, Q.; Luo, D.; Yao, B.; Tang, H.; et al. LCE: An open web portal to explore gene expression and clinical associations in lung cancer. Oncogene 2018, 38, 2551. [Google Scholar] [CrossRef] [PubMed]
- The American Cancer Society Medical and Editorial Content Team. What Is Non-Small Cell Lung Cancer. Available online: https://www.cancer.org/cancer/small-cell-lung-cancer/about/what-is-small-cell-lung-cancer.html (accessed on 4 June 2019).
- Sun, S.; Schiller, J.H.; Gazdar, A.F. Lung cancer in never smokers—A different disease. Nat. Rev. Cancer 2007, 7, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Richtmann, S.; Wilkens, D.; Warth, A.; Lasitschka, F.; Winter, H.; Christopoulos, P.; Herth, F.J.F.; Muley, T.; Meister, M.; Schneider, M.A. FAM83A and FAM83B as Prognostic Biomarkers and Potential New Therapeutic Targets in NSCLC. Cancers 2019, 11, 652. [Google Scholar] [CrossRef] [PubMed]
- Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 2010, 36, 1–48. [Google Scholar] [CrossRef]
- Schwarzer, G. Meta: An R package for meta-analysis. R. News 2007, 7, 40–45. [Google Scholar]
- Sergushichev, A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 2016. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lanczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef]
- Bravata, D.M.; Olkin, I. Simple pooling versus combining in meta-analysis. Eval. Health Prof. 2001, 24, 218–230. [Google Scholar] [CrossRef]
- Visbal, A.L.; Williams, B.A.; Nichols, F.C., 3rd; Marks, R.S.; Jett, J.R.; Aubry, M.C.; Edell, E.S.; Wampfler, J.A.; Molina, J.R.; Yang, P. Gender differences in non-small-cell lung cancer survival: An analysis of 4,618 patients diagnosed between 1997 and 2002. Ann. Thorac. Surg. 2004, 78, 209–215; discussion 215. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Larsen, J.E.; Lee, W.; Sun, H.; Shames, D.S.; Dalvi, M.P.; Ramirez, R.D.; Tang, H.; DiMaio, J.M.; Gao, B.; et al. Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Mol. Cancer Res. 2013, 11, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Xiao, G.; Behrens, C.; Schiller, J.; Allen, J.; Chow, C.W.; Suraokar, M.; Corvalan, A.; Mao, J.; White, M.A.; et al. A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clin. Cancer Res. 2013, 19, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Aerts, J.; den Hamer, B.; van Ijcken, W.; den Bakker, M.; Riegman, P.; van der Leest, C.; van der Spek, P.; Foekens, J.A.; Hoogsteden, H.C.; et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 2010, 5, e10312. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wistuba, I.I.; Behrens, C.; Virmani, A.K.; Mele, G.; Milchgrub, S.; Girard, L.; Fondon, J.W., 3rd; Garner, H.R.; McKay, B.; Latif, F.; et al. High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res. 2000, 60, 1949–1960. [Google Scholar]
- Angeloni, D. Molecular analysis of deletions in human chromosome 3p21 and the role of resident cancer genes in disease. Brief. Funct. Genom. Proteom. 2007, 6, 19–39. [Google Scholar] [CrossRef]
- Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; Misek, D.E.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar]
- Marinho, F.C.; Takagaki, T.Y. Hypercoagulability and lung cancer. J. Bras. Pneumol. 2008, 34, 312–322. [Google Scholar] [CrossRef]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef]
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Kamma, H.; Yazawa, T.; Ogata, T.; Horiguchi, H.; Iijima, T. Expression of MHC class II antigens in human lung cancer cells. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1991, 60, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Fukuyama, Y.; Yokoyama, H.; Kuninaka, S.; Asoh, H.; Katsuda, Y.; Ichinose, Y. HLA class I and class II expression of pulmonary adenocarcinoma cells and the influence of interferon gamma. Lung Cancer 1998, 20, 185–190. [Google Scholar] [CrossRef]
- He, Y.; Rozeboom, L.; Rivard, C.J.; Ellison, K.; Dziadziuszko, R.; Yu, H.; Zhou, C.; Hirsch, F.R. MHC class II expression in lung cancer. Lung Cancer 2017, 112, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Faruki, H.; Mayhew, G.M.; Serody, J.S.; Hayes, D.N.; Perou, C.M.; Lai-Goldman, M. Lung Adenocarcinoma and Squamous Cell Carcinoma Gene Expression Subtypes Demonstrate Significant Differences in Tumor Immune Landscape. J. Thorac. Oncol. 2017, 12, 943–953. [Google Scholar] [CrossRef]
- Bartel, C.A.; Parameswaran, N.; Cipriano, R.; Jackson, M.W. FAM83 proteins: Fostering new interactions to drive oncogenic signaling and therapeutic resistance. Oncotarget 2016, 7, 52597–52612. [Google Scholar] [CrossRef]
- Snijders, A.M.; Lee, S.Y.; Hang, B.; Hao, W.; Bissell, M.J.; Mao, J.H. FAM83 family oncogenes are broadly involved in human cancers: An integrative multi-omics approach. Mol. Oncol. 2017, 11, 167–179. [Google Scholar] [CrossRef]
- Lee, S.Y.; Meier, R.; Furuta, S.; Lenburg, M.E.; Kenny, P.A.; Xu, R.; Bissell, M.J. FAM83A confers EGFR-TKI resistance in breast cancer cells and in mice. J. Clin. Investig. 2012, 122, 3211–3220. [Google Scholar] [CrossRef]
- Cipriano, R.; Graham, J.; Miskimen, K.L.; Bryson, B.L.; Bruntz, R.C.; Scott, S.A.; Brown, H.A.; Stark, G.R.; Jackson, M.W. FAM83B mediates EGFR-and RAS-driven oncogenic transformation. J. Clin. Investig. 2012, 122, 3197–3210. [Google Scholar] [CrossRef]
- Okabe, N.; Ezaki, J.; Yamaura, T.; Muto, S.; Osugi, J.; Tamura, H.; Imai, J.; Ito, E.; Yanagisawa, Y.; Honma, R.; et al. FAM83B is a novel biomarker for diagnosis and prognosis of lung squamous cell carcinoma. Int. J. Oncol. 2015, 46, 999–1006. [Google Scholar] [CrossRef]
- Gainor, J.F.; Varghese, A.M.; Ou, S.H.; Kabraji, S.; Awad, M.M.; Katayama, R.; Pawlak, A.; Mino-Kenudson, M.; Yeap, B.Y.; Riely, G.J.; et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: An analysis of 1,683 patients with non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 4273–4281. [Google Scholar] [CrossRef] [PubMed]
- Kawase, A.; Yoshida, J.; Ishii, G.; Nakao, M.; Aokage, K.; Hishida, T.; Nishimura, M.; Nagai, K. Differences between squamous cell carcinoma and adenocarcinoma of the lung: Are adenocarcinoma and squamous cell carcinoma prognostically equal? Jpn. J. Clin. Oncol. 2012, 42, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.W.; Karakasheva, T.A.; Lee, D.J.; Lee, J.S.; Long, Q.; Bass, A.J.; Wong, K.K.; Rustgi, A.K. Comparative transcriptomes of adenocarcinomas and squamous cell carcinomas reveal molecular similarities that span classical anatomic boundaries. PLoS Genet. 2017, 13, e1006938. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, X.; Du, J.; Wang, X.J.; Xia, L. Differentiated regulation of immune-response related genes between LUAD and LUSC subtypes of lung cancers. Oncotarget 2017, 8, 133–144. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, L.; Luo, D.; Yao, B.; Yang, D.M.; Lin, S.; Girard, L.; DeBerardinis, R.J.; Minna, J.D.; Xie, Y.; Xiao, G. Systematic Analysis of Gene Expression in Lung Adenocarcinoma and Squamous Cell Carcinoma with a Case Study of FAM83A and FAM83B. Cancers 2019, 11, 886. https://doi.org/10.3390/cancers11060886
Cai L, Luo D, Yao B, Yang DM, Lin S, Girard L, DeBerardinis RJ, Minna JD, Xie Y, Xiao G. Systematic Analysis of Gene Expression in Lung Adenocarcinoma and Squamous Cell Carcinoma with a Case Study of FAM83A and FAM83B. Cancers. 2019; 11(6):886. https://doi.org/10.3390/cancers11060886
Chicago/Turabian StyleCai, Ling, Danni Luo, Bo Yao, Donghan M. Yang, ShinYi Lin, Luc Girard, Ralph J. DeBerardinis, John D. Minna, Yang Xie, and Guanghua Xiao. 2019. "Systematic Analysis of Gene Expression in Lung Adenocarcinoma and Squamous Cell Carcinoma with a Case Study of FAM83A and FAM83B" Cancers 11, no. 6: 886. https://doi.org/10.3390/cancers11060886
APA StyleCai, L., Luo, D., Yao, B., Yang, D. M., Lin, S., Girard, L., DeBerardinis, R. J., Minna, J. D., Xie, Y., & Xiao, G. (2019). Systematic Analysis of Gene Expression in Lung Adenocarcinoma and Squamous Cell Carcinoma with a Case Study of FAM83A and FAM83B. Cancers, 11(6), 886. https://doi.org/10.3390/cancers11060886