Abstract
In this study, we evaluated the utility of extracellular RNA (exRNA) derived from the plasma of multiple myeloma (MM) patients for whole transcriptome characterization. exRNA from 10 healthy controls (HC), five newly diagnosed (NDMM), and 12 relapsed and refractory (RRMM) MM patients were analyzed and compared. We showed that ~45% of the exRNA genes were protein-coding genes and ~85% of the identified genes were covered >70%. Compared to HC, we identified 632 differentially expressed genes (DEGs) in MM patients, of which 26 were common to NDMM and RRMM. We further identified 54 and 191 genes specific to NDMM and RRMM, respectively, and these included potential biomarkers such as LINC00863, MIR6754, CHRNE, ITPKA, and RGS18 in NDMM, and LINC00462, PPBP, RPL5, IER3, and MIR425 in RRMM, that were subsequently validated using droplet digital PCR. Moreover, single nucleotide polymorphisms and small indels were identified in the exRNA, including mucin family genes that demonstrated different rates of mutations between NDMM and RRMM. This is the first whole transcriptome study of exRNA in hematological malignancy and has provided the basis for the utilization of exRNA to enhance our understanding of the MM biology and to identify potential biomarkers relevant to the diagnosis and prognosis of MM patients.
1. Introduction
Multiple myeloma (MM) is a neoplasm of terminally differentiated plasma cells (PC), which at diagnosis is usually present throughout the bone marrow (BM) and frequently, with disease progression, spreads to extramedullary locations. The treatment of MM has witnessed significant progress with the introduction of proteasome inhibitors and immunomodulatory agents; however, the disease remains incurable with MM cells acquiring resistance to systemic therapies due to the accumulation of additional mutations that are often not present during the initial stages of the disease [1]. Resistance to therapy is mediated through the genetic evolution of MM cells under selective pressure treatment with more resistant clones emerging that possess growth and survival advantages [2].
Next-generation deep sequencing of MM tumor specimens has identified widespread genetic heterogeneity in MM [3,4]. Gene expression profiling (GEP) studies of PC isolated from patients presenting with the pre-MM precursor syndrome, monoclonal gammopathy of undetermined significance (MGUS), and symptomatic MM, have identified specific transcriptional changes that occur at different stages of the disease and these studies have reported pathways that had not been previously implicated in MM pathogenesis [5]. The current practice for obtaining genetic information about the tumor is to perform sequential BM biopsies and this is confounded by the known inter and intra-clonal spatial and genetic heterogeneity of the tumor(s) [1,2,6]. An alternative approach that we believe will provide a more comprehensive transcriptional picture is to analyze the extracellular RNA (exRNA) from the peripheral blood (PB) plasma, as this theoretically contains genetic material that arises not only from multiple independent MM tumors but also from the tumor microenvironment (TME).
Nucleic acids are released into the plasma and serum through cellular apoptosis, necrosis, and the spontaneous release of DNA/RNA-lipoprotein complexes, amongst other sources [7]. The presence of cell-free (cf) nucleic acids in the human blood was described more than 60 years ago [8]. A number of recent studies in breast, ovarian, lung, and colorectal cancers have indicated that tumor(s) from both primary and secondary sites release nucleic acids into the PB, as such, circulating cfDNA contains a representation of the entire genome of the tumor with DNA sourced from multiple independent tumors. Whole genome sequencing and whole exome sequencing of the cfDNA can be utilized to identify mutations associated with acquired resistance to cancer therapy without the need to perform sequential biopsies of the tumor [9,10]. Also, some studies have uncovered the presence of microRNAs and specific long noncoding RNAs in the exosomes, an extracellular vesicle subtype ranging from 30–150 nm in size [11], isolated from the blood of MM patients [12,13]. However, exRNA has not been systematically explored, with only a limited number of studies exploring the relevance of specific mRNA transcripts [14,15,16,17,18]. More recently, technologies have advanced that promote the reliable extraction and analysis of exRNA using high-throughput sequencing technologies, and a limited number of studies have also performed whole transcriptome studies of exRNA in pregnancy and Alzheimer’s disease [19,20,21]. However, to our knowledge, no studies have explored the whole transcriptome of exRNA in MM or other cancers.
Global gene expression profiling of PC from normal, MGUS, smoldering (asymptomatic) myeloma (SMM), and symptomatic MM patients has indicated that MGUS patients have a profile that is different to SMM and MM patients and that a gene-based classification system can be employed for MM [22,23,24]. In this study, we profiled the whole transcriptome of exRNA obtained from the PB plasma of healthy controls (HC) and MM patients to identify potential biomarkers for both newly diagnosed (NDMM) and relapsed and refractory (RRMM) MM patients.
2. Materials and Methods
2.1. Peripheral Blood (PB) Collection and Processing
Peripheral blood (PB) samples were obtained from healthy volunteers (HC) and MM patients using 10 mL Streck RNA blood collection tubes (BCTs) following informed consent as per the Alfred Hospital Human Research and Ethics Committee. Immediately upon sample collection, the tubes were inverted to mix the blood with the preservative in the collection tube. The patented preservative in Cell-Free RNA BCT preserves exRNA in plasma and prevents the release of non-target background RNA from blood cells during sample processing and storage [25,26]. The Streck RNA tubes allow the collection of high-quality stabilized exRNA for rare target detection and determining accurate exRNA concentrations. Streck RNA BCT tubes were stored at room temperature and processed for plasma collection within 24 h of PB collection. Plasma was separated from PB through centrifugation at 820× g for 10 min. The supernatant was collected without disturbing the cellular layer and centrifuged again at 16,000× g for 10 min to remove any residual cellular debris. The supernatant was collected and stored at −80 °C until exRNA extraction.
2.2. exRNA Extraction
Frozen plasma samples (6 mL) were used to extract the exRNA using the QIAamp circulating nucleic acid kit (Qiagen, Hilden, Germany), according to the manufacturers’ instructions. Subsequently, a Qubit 2.0 Fluorometer (Life Technologies, Victoria, Australia) was used to measure the RNA yield, according to the manufacturer’s instructions with freshly prepared RNA standards. The maximum input volume utilized for the QUBIT assay was 5 μL. The RNA concentration was quantified as being between 0.5–90 ng/μL. The extracted RNA was stored at −80 °C until further processing for RNA-Seq.
2.3. Transcriptome Library Construction and Sequencing
After the RNA sample was treated using the Ribo-Zero Gold rRNA Removal Kit (Illumina, San Diego, CA, USA), a total amount of 100 ng RNA was used for cDNA library construction using the TruSeq™ RNA Library Preparation Kit v2 protocol (Illumina, San Diego, CA, USA), as previously described [27]. Then, the amplified cDNA libraries were assessed for quality using both the Agilent 2100 Bioanalyzer and qRT-PCR. After primer ligation sequencing, qRT-PCR was again used for quantification, and the final library was sequenced on an Illumina HiSeq 2000 platform (paired-end 100 bp) at the Australian Genomics Research Facility (Melbourne, Australia).
2.4. Analysis of Transcriptome Data
Raw transcriptome sequencing data were cleaned using SOAPnuke under default parameters [28]. The clean reads were aligned to the human reference genome (ENSEMBL, GRCH38.85) using Hisat2 and the expression of protein-coding and noncoding genes were profiled using Stringtie [29]. HTSeq (v0.9.0, https://htseq.readthedocs.io) was used to obtain the raw read counts aligned to each gene.
The TPM (transcripts per million reads) method was used to normalize the gene expression in each sample, and lowly expressed (<5 TPM) genes were filtered. To perform differential expression analysis, we first removed individual differences among samples by filtering genes with high coefficient values (>0.8) of standard variation in each group. An R package named as edgeR was used to calculate the statistical values for differentially expressed genes (DEGs) [30] and the DEGs were identified based on the following criteria: TPM > 5, log2 fold change (Log2FC) > 1 or Log2FC < −1, p-value < 0.05 and FDR < 0.05. Principle component analysis was performed using the R package “prcomp”. Genome alignment files could be accessed on the National Center for Biotechnology Information (NCBI) platform under the accession number SRA893057.
2.5. Functional Analysis
Functional analysis of DEGs was performed using the DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov).
2.6. Identification of SNP and Indel Variations
Variants including SNPs and indels were called using the Genome Analysis Toolkit (GATK v3.4) pipeline, according to the Best Practice workflow. In brief, the clean reads for each sample were aligned to the human reference genome (GRCH38) using the STAR 2-pass alignment steps [31]. Then, the resulting SAM file was put through the usual Picard processing steps, including adding read group information, sorting, marking duplicates, and indexing (http://broadinstitute.github.io/picard/). Next, the RNA-Seq specific GATK tool called SplitNCigarReads was used to split the reads into exon segments and hard-clip any sequences overhanging into the intronic regions. After the indel realignment and base recalibration, we used HaplotypeCaller to call SNPs and indels, followed by variant filtering with recommended commands and parameters. We further filtered the results by removing the variants detected in less than half of the sample size. Final variants in MM patients excluded the events detected in HC. ANNOVAR was used to annotate the variants [32].
2.7. Droplet Digital PCR
We randomly selected 10 samples used for RNA-Seq, including three HC, two NDMM, and five RRMM patients for gene expression validation using droplet digital PCR (ddPCR). Initially, 3 mL of plasma samples were used for exRNA extraction, as described. Then, a NanoDrop 1000 was used to quantify the RNA yield. One-Step RT-ddPCR Advanced Kit for Probes (Bio-Rad Laboratories, Hercules, CA, USA) was used for the detection of five DEGs, including RGS18 (dHsaCPE5054808), ITPKA (dHsaCPE5192800), PPBP (dHsaCPE5039499), FTH1 (dHsaCPE5031151), and IER3 (dHsaCPE5190327), according to the manufacturer’s instructions. Briefly, the reaction mix (22 µL) was prepared with 5 µL Supermix (1×), 2 µL reverse transcriptase (20 U/µL), 1 µL DTT (300 nM), 1 µL gene probe (900 nM), 1 µL total RNA, and 12 µL RNase-free water. Then, the droplets were generated on the Automated Droplet Generator (Bio-Rad), followed by the thermal cycling reaction. The cycling conditions were conducted on a C1000 Touch Thermal Cycler (Bio-Rad) as follows: reverse transcription (46 °C, 60 min), enzyme activation (95 °C, 10 min), 40 cycles of denaturation (95 °C, 30 s), and annealing/extension (60 °C, 1 min), and enzyme deactivation (98 °C, 10 min). After thermal cycling, the plates were read by the QX200 Droplet Reader (Bio-Rad) and the QuantaSoft software was used to analyze the data. To reduce the errors, we employed GAPDH (Glyceraldehyde-3-Phosphate Dehydrogenase) as an internal control and the gene expression was shown relative to the absolute copies of GAPDH. Aberrant values (>500 copies/µL) were set to null, and samples with no gene expression were not counted for the calculation of the average gene expression.
3. Results
3.1. Characteristics of MM Patients and RNA Profiles
To investigate the biomarker potential of exRNA for NDMM and RRMM, this study recruited a total of 27 volunteers, including 10 HC (six males and four females), five NDMM (three males and two females) patients, and 12 RRMM (seven males and five females) patients. No difference was found between the groups based on gender, moreover, as shown in Table 1, the mean age of NDMM (66 years) and RRMM (64.6 years) were also similar (older than the HC group: 41.9 years). After the total RNA was extracted, we evaluated the RNA profiles of exRNA using an Agilent Bioanalyzer 2100. Unlike the RNA profiles of whole cell lysates, the exRNA profiles lacked the 18S and 28S rRNA peaks (Supplementary Figure S1). Whether this is one of the characteristics of exRNA requires further experiments to explore because in this instance, the exRNA sample was treated with the Ribo-Zero Gold rRNA Removal Kit.

Table 1.
Description of participants in this study. No bias was found in age or sex in the participants for each group. HC: healthy control; ND: newly diagnosed MM patient; RR: relapsed and refractory MM patient; IgA: immunoglobulin A; FISH: fluorescence in situ hybridization. Translocation t(4:14) represents the FGFR3-IGH fusion event while t(14:16) represents the IGH-MAF fusion event.
3.2. Overview of the Transcriptome Sequencing
After low-quality reads, adapter and contamination reads were filtered by SOAPnuke; we obtained a total of 762.50 million paired reads (28.24 million reads per sample on average) (Table 2). The lowest Q20 and Q30 of this sequencing were 93.83% and 86.53%, respectively (Table 2). Using Hisat2, 88.40–96.78% of the clean reads were aligned to the human reference genome (GRCH38), and 83.20–94.73% of the clean reads were paired matched (Table 2). Then, we used Stringtie to profile the gene expression in each sample. As shown in Table 2, 23,274 to 38,293 genes were identified in the MM patients and HC, respectively. We next evaluated the coverage of identified genes in each sample and found that 79.76–88.58% of the identified genes were covered more than 70% by the sequencing reads (Figure 1A). Gene annotation showed that 38.71–50.51% of the identified genes had the potential of protein-coding capacity while 1.77–2.33% of the identified genes were to be experimentally confirmed (TEC) (Figure 1B). Three long-noncoding RNAs, including RN7SL1, RN7SL2, and RN7SL4P, were the most highly expressed genes in the HC, NDMM, and RRMM samples, followed by the Metazoa_SRP (Metazoan signal recognition particle RNA).
Table 2.
Overview of the extracellular RNA (exRNA) transcriptome sequencing. Q20 and Q30 were applied to the clean reads. The number of mapped reads was given by Hisat2 and ‘.5’ means that one of the paired reads was mapped. Percentages were calculated for mapped reads and paired reads matched to the genome. The numbers of genes identified in the samples (>5 transcripts per million reads (TPM)) were listed.
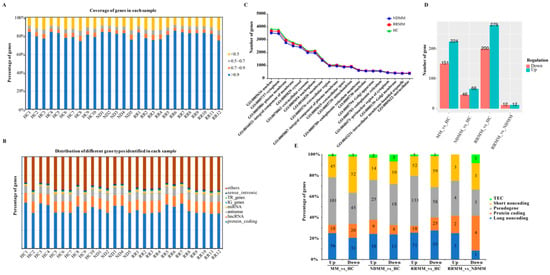
Figure 1.
Overview of the transcriptome of extracellular RNA (exRNA) in healthy controls (HC) and multiple myeloma (MM) patients. (A) Distribution of gene coverage in each sample. (B) Distribution of gene types in each sample. (C) Top 20 terms of the GO cellular component identified by the genes in HC, newly diagnosed multiple myeloma (NDMM), and relapsed and refractory multiple myeloma (RRMM). (D) Status of differentially expressed genes identified in MM compared to HC. (E) Distribution of gene types for the differentially expressed genes (DEGs).
We next analyzed the gene ontology in terms of the cellular component to source the exRNA molecules in cells. The top 20 enriched cellular components of GO were shared by NDMM, RRMM, and HC (Figure 1C). With the exception of the nucleus and the cytoplasm, the genes were enriched in the membrane and extracellular vesicle-related GO terms, such as the “plasma membrane (GO:0005886)” and “extracellular exosome (GO:0070062)”.
3.3. Dysregulated exRNA in MM Patients
To identify potential exRNA biomarkers for NDMM and RRMM, we performed differential gene expression analysis using edgeR under the criteria described in the methods. First, we compared all the MM (NDMM + RRMM) patients to the HC and identified 375 DEGs (224 up-regulated and 151 down-regulated) (Figure 1D, Supplementary Table S1). As shown in Figure 1E, 38 protein-coding genes (18 up-regulated and 20 down-regulated), 87 long noncoding genes (56 up-regulated and 31 down-regulated), 97 short noncoding genes (45 up-regulated and 52 down-regulated), and seven TEC genes (four up-regulated and three down-regulated) were differentially expressed in MM compared to HC. Apart from the immunoglobulin-related genes, GOLGA8O (golgin A8 family member O) and KCTD14 (potassium channel tetramerization domain containing 14) were the top two up-regulated genes, while the top two down-regulated genes were C21orf33 (chromosome 21 open reading frame 33) and RPL13A (ribosomal protein L13a).
Next, compared to HC, we identified 66 up-regulated and 46 down-regulated genes (Figure 1D, Supplementary Table S1) in NDMM, including 13 protein-coding genes (nine up-regulated and four down-regulated), 27 long noncoding genes (16 up-regulated and 11 down-regulated), 43 pseudogenes (25 up-regulated and 18 down-regulated), 24 short noncoding genes (14 up-regulated and 10 down-regulated), and five TEC (two up-regulated and three down-regulated) (Figure 1E). The nine up-regulated protein-coding genes were MYOD1, AC009060.2, CHRNE, GOLGA8O, NKX2-4, FTH1, UBB, OLIG1, and ITPKA. The four down-regulated protein-coding genes were RGS18, HISTH1B, TRK2, and MBD3L5.
In RRMM, we identified 279 up-regulated and 200 down-regulated genes compared to HC (Figure 1D, Supplementary Table S1), including 43 protein-coding genes (18 up-regulated and 25 down-regulated), 126 long noncoding genes (71 up-regulated and 55 down-regulated), 191 pseudogenes (133 up-regulated and 58 down-regulated), 111 short noncoding genes (52 up-regulated and 59 down-regulated), and eight TEC (five up-regulated and three down-regulated) (Figure 1E). The highly up-regulated protein-coding genes included immunoglobulin genes and GOLGA8O, while the highly down-regulated protein-coding genes included HIST1H4A and RPL13A.
3.4. Potential exRNA Biomarkers for MM Patients
A heat map (Figure 2A) showed that DEGs could be distinguished between MM patients and HC. This was also revealed by the principal component analysis (Figure 2B). However, due the limited sample size of the NDMM, it was difficult to separate NDMM from RRMM (Figure 2B). We next compared the DEGs identified in NDMM and RRMM. As shown in Figure 2C, 18 up-regulated and eight down-regulated genes were common to MM (NDMM + RRMM), including one up-regulated protein-coding gene (GOLGA8O) and one down-regulated protein-coding gene (TRAK2). We also identified 48 up-regulated and 38 down-regulated genes specific to NDMM (Figure 2C, Supplementary Table S1), which could be utilized as potential biomarkers for NDMM. The NDMM specific up-regulated genes included seven lincRNA genes (e.g., AC007405.6, LINC00863, RP11-573G6.10, and RP11-626H12.1), one primary miRNA gene (MIR6754), and eight protein-coding genes (e.g., MYOD1, AC009060.2, CHRNE, NKX2-4, FTH1, UBB, OLIG1, and ITPKA). The NDMM specific down-regulated genes included five lincRNA genes (e.g., PACERR, LINC01123, RP11-596C23.2, and AC137934.1) and three protein-coding genes (e.g., MBD3L5, RGS18 and HIST1H1B). In addition, we identified 261 up-regulated and 192 down-regulated genes specific to RRMM (Figure 2C, Supplementary Table S1). The RRMM specific dysregulated exRNA genes included 34 protein-coding genes (11 up-regulated and 23 down-regulated), 52 lincRNA genes (33 up-regulated and 19 down-regulated), 18 primary miRNA genes (10 up-regulated and eight down-regulated), and 37 misc_RNA genes (18 up-regulated and 19 down-regulated).
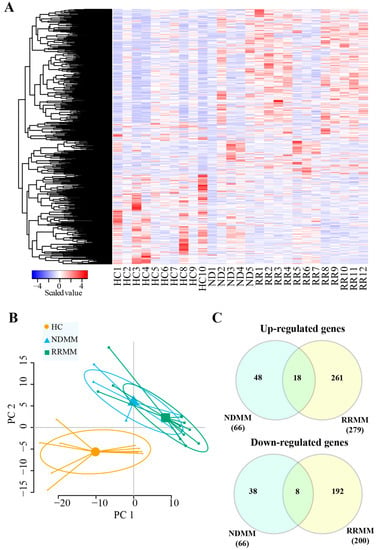
Figure 2.
Comparison of DEGs in NDMM and RRMM. (A) A heat map of the DEGs showed different signatures for HC, NDMM, and RRMM. (B) Principle component analysis of all samples. (C) Venn diagrams of up- and down-regulated genes identified in all MM patients, NDMM, and RRMM.
Next, we compared the exRNA profiles of NDMM and RRMM. Using edgeR we identified a total of 24 genes differentially expressed in RRMM relative to NDMM, including 12 up-regulated and 12 down-regulated genes (Figure 1D). The distribution of different gene types in the DEGs of RR and ND (Figure 1E) showed that six protein-coding DEGs, including two up-regulated genes (IGKV1-5 and OR4N4) and four down-regulated genes (AC017081.1, UBB, CHRNE, and USP17L17) were dysregulated in RRMM compared to NDMM. Among them, UBB and CHRNE were up-regulated in NDMM and UBB was also up-regulated in MM (NDMM + RRMM) (Supplementary Table S1). Also, we found a lincRNA gene AC137934.1 up-regulated in RRMM compared to NDMM but down-regulated in NDMM compared to HC.
Functional analysis using DAVID bioinformatics resources showed that 10 and 11 DEGs in RRMM compared to HC were involved in the biological process of “SRP-dependent co-translational protein targeting to membrane” (GO:0006614) and “Ribosome” (hsa03010) pathways, respectively. Also, two DEGs in NDMM compared to HC were involved in the molecular function of “chromatin DNA binding” (GO:0031490). According to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation, we also found that ITPKA, which was up-regulated in NDMM, was related to signal transduction. Among the DEGs in the RRMM, PPBP (down-regulated) and CCL4L2 (up-regulated) were involved in the “chemokine signaling pathway” (ko04062), HIST1H4A (down-regulated) was related to “Viral carcinogenesis” (ko05203), and PCBP1 (down-regulated) was a regulator of “spliceosome” (ko03040).
3.5. Gene Variants in MM Patients
We next used the GATK pipeline to detect SNPs and small indels in MM patients. As shown in Figure 3A, we first identified 3,606,315 SNPs in HC, of which 79,008 were found in more than five individuals. We also identified 97,547 SNPs in more than two NDMM and 159,506 SNPs in more than six RRMM. The SNPs identified in HC were classified as background and were filtered from the MM patients, resulting in 734,79 and 125,399 SNPs in NDMM and RRMM, respectively. There were 19,320 SNPs shared between NDMM and RRMM (Figure 3A). ANNOVAR was used to annotate the SNPs, and the results showed that 137 exonic SNPs were identified in both NDMM and RRMM, including one splicing event, 80 nonsynonymous, and 57 synonymous single-nucleotide variants (SNVs) (Figure 3A). The shared nonsynonymous SNPs all have been annotated in dbSNP (Table 3). Seven SNPs were identified in more than four NDMM patients and more than 10 RRMM patients, including rs61821060, rs2363468, rs1941635, rs2172521, rs7199961, rs73714227, and rs4728137. rs61821060 is a mutation of the RPFIA4 gene (chr1 203039046, G->C) and was found in five NDMM and 11 RRMM patients. There were 388 and 861 exonic SNPs specific to NDMM and RRMM, respectively (Figure 3A). The 388 NDMM specific exonic SNPs included 168 nonsynonymous (Supplementary Table S2), 219 synonymous, and one stop-loss SNP. Notably, we found one nonsynonymous SNP (chr7, 100958135, T->C, MUC3A) that has not been reported in the Single Nucleotide Polymorphism Database (dbSNP). The 861 RRMM specific exonic SNPs included 403 nonsynonymous (Supplementary Table S3), 446 synonymous, two stop-gain and 10 unknown SNPs. Of the 403 nonsynonymous SNPs we identified five novel mutations, two occurred in MUC5AC (chr11, 1191524, T->C; chr11, 1191527, C->T), and three in MUC3A (chr7, 100953992, C->A; chr7, 100954123, G->A; chr7, 100954136, C->G). Next, we analyzed the mutations occurring in intestinal mucin genes, such as MUC3A, MUC5AC, MUC12 and MUC16. It was revealed that more SNPs occurred in mucin genes in RRMM than in NDMM (Figure 3B), particularly the MUC5AC gene.
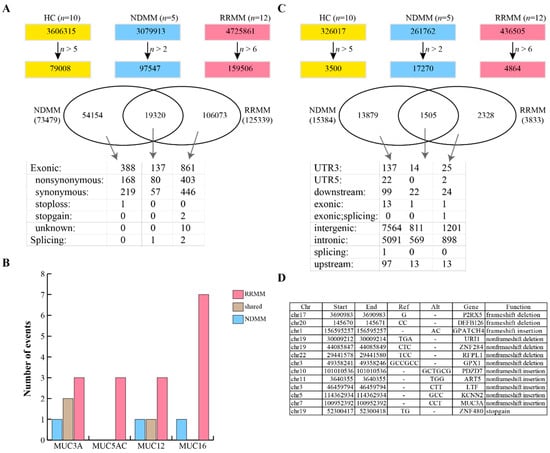
Figure 3.
SNP and indel analysis. (A) Identification of SNPs in MM and their annotation. (B) Comparison of the distribution and the frequency of mucin gene mutations in NDMM and RRMM. (C) Identification of small indels in NDMM and RRMM. (D) Annotation of 13 known small indels specific to NDMM.
Table 3.
Common nonsynonymous SNPs identified in NDMM and RRMM. The number of NDMM and RRMM patients were listed.
We next evaluated for the presence of indels in the MM patients. As shown in Figure 3C, a total of 15,384 and 3833 indels were identified in NDMM and RRMM, respectively. NDMM and RRMM shared 1505 indels, of which one was derived from the exonic region. The shared exonic indel was annotated as rs11402251, which is located in the 10th exon of the VSIG10L gene and is a frameshift insertion. Two known indels (rs5843453 and rs55745992) were identified specifically in RRMM, while 13 known indels were identified specifically in NDMM (Figure 3D). Interestingly, nine out of the 13 NDMM specific indels had no frameshift function to their mother genes. The left indels included frameshift deletions on P2RX5 and DEFB126, frameshift insertions to GPATCH4, and a stop-gain insertion to ZNF480 (Figure 3D).
Functional analysis by DAVID showed that mutated genes for both NDMM and RRMM were involved in “Olfactory transduction (hsa04740)” and “GO:0007186~G-protein-coupled receptor signaling pathway”. In addition, RRMM specific mutated genes were also involved in “ECM-receptor interaction (hsa04512)”, “GO:0007155~cell adhesion”, “GO:0071813~lipoprotein particle binding”, and “PI3K-AKT signaling pathway (hsa04151)”.
3.6. Digital Droplet PCR Validation
We next utilized ddPCR to validate five DEGs (RGS18, ITPKA, PPBP, FTH1, and IER3) in the exRNA samples from 10 participants (three HC, two NDMM, and five RRMM). We used relative normalized expression (RNE, relative to GAPDH) to present the gene expression detected by ddPCR. Figure 4 showed the comparison of gene expression identified by ddPCR and RNA-Seq. Among them, the expression levels of three genes (RGS18, ITPKA, and PPBP) were consistent between these two experiments in HC, NDMM, and RRMM. In addition, FTH1 was up-regulated in NDMM compared to HC (Log2FC = 1.78, FDR = 2.16 × 10−7, Supplementary Table S1) and its up-regulation was also confirmed by ddPCR. The abnormal performance of FTH1 in Figure 4 is due to its high expression in the three HCs. The disagreement of ddPCR and RNA-Seq on the expression change of IER3 require further experiments for validation.
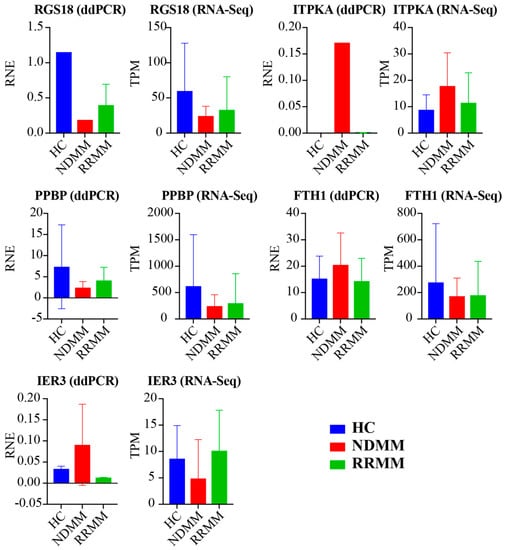
Figure 4.
Digital Droplet PCR (ddPCR) validation. Five DGEs were selected and compared to RNA-Seq. Their expression was normalized to GAPDH and was present in relative normalized expression (RNE) by ddPCR. Their normalized expression identified by RNA-Seq was shown in transcripts per million reads (TPM).
4. Discussion
In this study, we evaluated plasma-derived exRNA from MM patients and healthy individuals (Table 2). The exRNA has been reported to be subjected to degradation, instability, low abundance, and intracellular communication from specimen processing [33,34]; however, in our study we demonstrated that 79.76–88.58% of the identified genes were covered more than 70%, which means that a large set of gene transcripts was complete in the exRNA tested. The stability of exRNA in the plasma or serum may be due to the protection of extracellular vesicles, such as exosomes, microparticles, microvesicles, or multivesicles [35], which are shed from cellular surfaces into the bloodstream [11]. In support of this, we observed that the RNA profiles generated by the Agilent Bioanalyzer 2100 were similar to exosomal RNA profiles [36], which lack 18S and 28S rRNA peaks. Furthermore, our results showed that 2002–2135 genes were related to the “extracellular exosome (GO:0070062)” (Figure 1C). New evidence has shown that a large proportion of human blood plasma cf-DNA is localized in exosomes [37]; however, the proportion of exosomal RNA in exRNA requires further investigation.
No studies have investigated the global transcriptome profiles of exRNA in MM. In this study, we identified genes in exRNA that could potentially be used as diagnostic and prognostic biomarkers for MM, including 18 up-regulated and eight down-regulated genes (Figure 2B, Supplementary Table S1). GOLGA8O was the only up-regulated protein-coding gene common to both NDMM and RRMM. It has been reported to be significantly down-regulated in patients with major depressive disorder [38]; however, the function of this gene in cancers has not been investigated. The common down-regulated genes in MM included TRAK2, a possible regulator of endosome-to-lysosome trafficking of membrane cargo, including the epidermal growth factor receptors (EGFR) [39]. Endosome-to-lysosome trafficking is also an important process in exosome biogenesis [11]. In addition, TRAK1, the ortholog of TRAK2, has been identified as MGb2-Ag—a novel cancer biomarker [40]. In MM, the down-regulation of TRAK2 might be due to the up-regulation of miR-19a [41,42], which is a member of miR-17~92a cluster and can target the TRAK2 gene [41].
In this study, we identified 54 genes specifically dysregulated in NDMM, including MYOD1, UBB, MIR6754, and PACERR (Supplementary Table S1). The hypermethylation of MYOD1 has been reported to be a prognostic biomarker for both colorectal and cervical cancers [43,44]. The polyubiquitin gene UBB, encoding a regulatory protein involved in ubiquitin, has been identified to be up-regulated more than 100-fold in MM patient cells versus normal twin plasma cells [45]. Moreover, the knockdown of UBB can significantly decrease the expression of ubiquitin, which is essential for cancer cell growth. UBB may, therefore, represent a potential target in anticancer treatment including in MM [46]. miR-6754 has been related to cell proliferation and invasion in non-small cell lung cancer [47]. PACERR is the antisense gene of PGTS2 (prostaglandin-endoperoxide synthase 2) that has been related to colorectal cancer and breast cancer [48,49]. There are currently no published studies that relate MIR6754 and PACERR or other noncoding RNAs to MM; however, their expression patterns in NDMM suggest that they might play a role in the progression of MM and may also represent potential biomarkers for MM.
Among the 191 genes specifically dysregulated in RRMM patients, seven down-regulated (PPBP, EEF1B2, RPL5, RPL19, CH507-9B2.3, EEF2, and RPS27) and three up-regulated (VN1R1, IER3 and POTED) protein-coding genes were identified (Supplementary Table S1). Studies have reported the overexpression of most of these genes in multiple cancers, with the exception of CH507-9B2.3 [50,51,52,53,54]; however, they have also been found to be down-regulated in some specific cancers. For example, the chemokine PPBP (pro-platelet basic protein), also known as CXCL7, is decreased in pancreatic and ovarian cancers [55]. The eukaryotic translation elongation factors EEF1B2 and EEF2 are down-regulated in breast, esophageal, and lung cancers while EEF1B2 is found with repression in some other cancer types, such as head and neck, leukemia, and pancreatic cancer [54]. Whether the down-regulation of these genes is specific to MM requires further study. Only three protein-coding genes were specifically up-regulated in RRMM, including VN1R1, POTED, and IER3 (Supplementary Table S1). Among them, VN1R1 has been shown to be overexpressed in prostate adenocarcinomas and glioblastoma cancer cells [56,57] and POTED in prostate cancer patients, making it a potential molecule for targeted therapy [58]. Interestingly, IER3 has been shown in pancreatic ductal adenocarcinoma to effectively mitigate against cellular stress induced by starvation or exposure to gemcitabine [59]. Notably, IER3 has also been demonstrated to have an anti-apoptotic role in MM endothelial cells and is overexpressed in MM plasma cells [60].
In comparison to protein-coding genes, a larger proportion of noncoding DEGs were up-regulated in MM (Supplementary Table S1), which is consistent with a previous study, albeit of PC-derived RNA [61]. Some lncRNAs are considered to be potential promoters of MM progression, and thus, could have potential as diagnostic and prognostic biomarkers, these include LINC01215 [62], MIR222HG [62], MEG3 [63], MALAT [64], CRNDE [65], and H19 [66]. In this study, additional lncRNAs with biomarker potential in MM were identified (Supplementary Table S1), including 65 antisense genes (e.g., FAM83C-AS1, ZNF32-AS1, TMC3-AS1, and TAT-AS1), 71 lincRNA genes (e.g., LINC00863, LINC01123, LINC00349, LINC00677, and LINC00462) and 25 primary miRNA genes (e.g., MIR301A, MIR378H, MIR425, and MIR647). Among these, some have been reported in other cancers, such as LINC01123 in prostate cancer [67], LINC00677 in acute myeloid leukemia [68] and LINC00462 in pancreatic cancer [69]. The function of these lncRNAs in MM and their diagnostic and prognostic potential requires further evaluation.
In this study, we also identified gene variants using the transcriptome data (Figure 3). Of interest were the RRMM specific indels related to both cell adhesion and PI3K-AKT signaling pathways. Unfortunately, the hotspot mutations on KRAS, NRAS, and TP53 genes, which are common in MM [70], were not identified in this study, probably due to the low coverage of these genes [71]. However, our findings revealed that mucin genes, such as MUC3A, MUC5AC, MUC12, and MUC16, may play a role in MM progression, as they were frequently mutated in the MM patient, particularly the RRMM patients. Moreover, MUC16 mutations, the most frequently mutated in this analysis, have been previously demonstrated to be associated with a higher tumor mutational burden and superior survival outcomes in gastric cancer patients [72]. Additionally, uterine endometrioid endometrial adenocarcinoma harbor a high-frequency of the MUC3A mutations [73]. Whether mucin gene variants are derived from MM is still unknown, and our results indicate that mucin genes might be related to the MM pathogenesis. These results demonstrate the potential capacity of exRNA interrogation to identify genetic variants; however, we acknowledge that more sequencing data (high-coverage) and DNA sequencing are required to validate the genomic mutations of the described mucin genes and more patient samples are needed for large scale scanning.
5. Conclusions
In conclusion, this is the first transcriptome study of exRNA in cancer and in our hands, we have demonstrated that ~85% of genes in exRNA were covered more than 70% and that ~45% of exRNA genes were protein-coding genes. DEGs identified in MM patients, including GOLGA8O and TRAK2 may be potential biomarkers for the disease. Importantly, we also identified specific differentially expressed protein-coding genes in both NDMM and RRMM including cancer-associated genes such as MYOD1, UBB, VN1R1, POTED, and IER3, and while they may have potential as biomarkers also warrants further study to determine their potential role in the pathogenesis of the disease. In addition, a range of nonsynonymous mutations were identified in the exRNA, including multiple mutated mucin genes and their role in MM also warrants further evaluation. While preliminary, these data demonstrate that exRNA may represent a valuable, non-invasive compartment not only for the identification of new diagnostic and prognostic biomarkers in MM but also for the study of the biology of the disease.
Supplementary Materials
The following are available online at https://www.mdpi.com/2072-6694/11/6/887/s1, Figure S1: Agilent 2100 Bioanalyzer results showed the absence of 18S and 28S rRNA in extracellular RNA (exRNA), Table S1: Differentially Expressed Genese (DEGs) identified in multiple myeloma (MM) patients compared to healthy controls (HC), Table S2: Non-synonymous SNPs identified only in newly diagnosed multiple myeloma (NDMM), Table S3: Non-synonymous SNPs identified only in relapsed and refractory multiple myeloma (RRMM).
Author Contributions
M.C., S.M. and A.S. designed the study. S.M., M.R., K.C., T.K. and M.C. collected the samples and conducted the experiments. S.M., T.K. and M.C. analyzed the experimental data and sequencing data. M.C., S.M. and A.S. wrote the manuscript, and all the authors have reviewed and approved the manuscript.
Funding
This study was funded by the International Myeloma Foundation’s Black Swan Research Initiative.
Acknowledgments
We would like to thank the participants in this study and all the staff from the Malignant Haematology & Stem Cell Transplantation, Alfred Hospital.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Egan, J.B.; Kortuem, K.M.; Kurdoglu, A.; Izatt, T.; Aldrich, J.; Reiman, R.; Phillips, L.; Baker, A.; Shi, C.X.; Schmidt, J.; et al. Extramedullary myeloma whole genome sequencing reveals novel mutations in Cereblon, proteasome subunit G2 and the glucocorticoid receptor in multi drug resistant disease. Br. J. Haematol. 2013, 161, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Egan, J.B.; Shi, C.X.; Tembe, W.; Christoforides, A.; Kurdoglu, A.; Sinari, S.; Middha, S.; Asmann, Y.; Schmidt, J.; Braggio, E.; et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012, 120, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Davies, F.E.; Dring, A.M.; Li, C.; Rawstron, A.C.; Shammas, M.A.; O’Connor, S.M.; Fenton, J.A.; Hideshima, T.; Chauhan, D.; Tai, I.T.; et al. Insights into the multistep transformation of MGUS to myeloma using microarray expression analysis. Blood 2003, 102, 4504–4511. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Sirdesai, S.; Chen, M.; Khong, T.; Spencer, A. Circulating Tumour DNA Analysis for Tumour Genome Characterisation and Monitoring Disease Burden in Extramedullary Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 1858. [Google Scholar] [CrossRef] [PubMed]
- Gahan, P.B.; Swaminathan, R. Circulating nucleic acids in plasma and serum. Recent developments. Ann. N. Y. Acad. Sci. 2008, 1137, 1–6. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Les acides nucleiques du plasma sanguin chez l’homme. Seances Soc. Biol. Ses Fil. 1948, 142, 241–243. [Google Scholar]
- Heitzer, E.; Auer, M.; Hoffmann, E.M.; Pichler, M.; Gasch, C.; Ulz, P.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Mohan, S.; et al. Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer. Int. J. Cancer 2013, 133, 346–356. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer—Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Liu, C.J.; Avet-Loiseau, H.; Park, J.; Shi, J.; Campigotto, F.; Salem, K.Z.; Huynh, D.; Glavey, S.V.; Rivotto, B.; et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 2017, 129, 2429–2436. [Google Scholar] [CrossRef] [PubMed]
- Sedlarikova, L.; Bollova, B.; Radova, L.; Brozova, L.; Jarkovsky, J.; Almasi, M.; Penka, M.; Kuglik, P.; Sandecka, V.; Stork, M.; et al. Circulating exosomal long noncoding RNA PRINS-First findings in monoclonal gammopathies. Hematol. Oncol. 2018, 36, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Joosse, S.A.; Muller, V.; Steinbach, B.; Pantel, K.; Schwarzenbach, H. Circulating cell-free cancer-testis MAGE-A RNA, BORIS RNA, let-7b and miR-202 in the blood of patients with breast cancer and benign breast diseases. Br. J. Cancer 2014, 111, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhang, J.; Sun, P.; Shang, J. Circulating cell-free human telomerase reverse transcriptase mRNA in plasma and its potential diagnostic and prognostic value for gastric cancer. Int. J. Clin. Oncol. 2013, 18, 478–486. [Google Scholar] [CrossRef] [PubMed]
- March-Villalba, J.A.; Martinez-Jabaloyas, J.M.; Herrero, M.J.; Santamaria, J.; Alino, S.F.; Dasi, F. Cell-free circulating plasma hTERT mRNA is a useful marker for prostate cancer diagnosis and is associated with poor prognosis tumor characteristics. PLoS ONE 2012, 7, e43470. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Wang, L.; Zheng, G.; Du, L.; Yang, Y.; Dong, Z.; Liu, Y.; Qu, A.; Wang, C. Investigation of cell free BIRC5 mRNA as a serum diagnostic and prognostic biomarker for colorectal cancer. J. Surg. Oncol. 2014, 109, 574–579. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Wang, L.; Du, L.; Wang, S.; Zheng, G.; Li, W.; Zhuang, X.; Zhang, X.; Dong, Z. Detection of circulating Bmi-1 mRNA in plasma and its potential diagnostic and prognostic value for uterine cervical cancer. Int. J. Cancer 2012, 131, 165–172. [Google Scholar] [CrossRef]
- Spornraft, M.; Kirchner, B.; Haase, B.; Benes, V.; Pfaffl, M.W.; Riedmaier, I. Optimization of extraction of circulating RNAs from plasma--enabling small RNA sequencing. PLoS ONE 2014, 9, e107259. [Google Scholar] [CrossRef]
- Zwemer, L.M.; Hui, L.; Wick, H.C.; Bianchi, D.W. RNA-Seq and expression microarray highlight different aspects of the fetal amniotic fluid transcriptome. Prenat. Diagn. 2014, 34, 1006–1014. [Google Scholar] [CrossRef]
- Koh, W.; Pan, W.; Gawad, C.; Fan, H.C.; Kerchner, G.A.; Wyss-Coray, T.; Blumenfeld, Y.J.; El-Sayed, Y.Y.; Quake, S.R. Noninvasive in vivo monitoring of tissue-specific global gene expression in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7361–7366. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Corral, L.; Corchete, L.A.; Sarasquete, M.E.; Mateos, M.V.; Garcia-Sanz, R.; Ferminan, E.; Lahuerta, J.J.; Blade, J.; Oriol, A.; Teruel, A.I.; et al. Transcriptome analysis reveals molecular profiles associated with evolving steps of monoclonal gammopathies. Haematologica 2014, 99, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Hardin, J.; Kordsmeier, B.; Bumm, K.; Zheng, M.; Tian, E.; Sanderson, R.; Yang, Y.; Wilson, C.; Zangari, M.; et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 2002, 99, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.S.; Li, Y.; Mitra, A.K.; Bi, L.; Abyzov, A.; van Wijnen, A.J.; Baughn, L.B.; Van Ness, B.; Rajkumar, V.; Kumar, S.; et al. Molecular signatures of multiple myeloma progression through single cell RNA-Seq. Blood Cancer J. 2019, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Norton, S.E.; Alt, J.R.; Krzyzanowski, G.D.; Williams, T.L.; Fernando, M.R. Stabilization of Cellular RNA in Blood During Storage at Room Temperature: A Comparison of Cell-Free RNA BCT((R)) with K3EDTA Tubes. Mol. Diagn. Ther. 2014, 18, 647–653. [Google Scholar] [CrossRef]
- Qin, J.; Williams, T.L.; Fernando, M.R. A novel blood collection device stabilizes cell-free RNA in blood during sample shipping and storage. BMC Res. Notes 2013, 6, 380. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xu, R.; Ji, H.; Greening, D.W.; Rai, A.; Izumikawa, K.; Ishikawa, H.; Takahashi, N.; Simpson, R.J. Transcriptome and long noncoding RNA sequencing of three extracellular vesicle subtypes released from the human colon cancer LIM1863 cell line. Sci. Rep. 2016, 6, 38397. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Li, X.; Zhang, F.; Zhang, C.; Guan, Q.; Cao, X.; Zhu, W.; Zhang, X.; Cheng, Y.; Ou, K.; et al. Circulating lncRNAs associated with occurrence of colorectal cancer progression. Am. J. Cancer Res. 2015, 5, 2258–2265. [Google Scholar] [PubMed]
- Yuan, T.; Huang, X.; Woodcock, M.; Du, M.; Dittmar, R.; Wang, Y.; Tsai, S.; Kohli, M.; Boardman, L.; Patel, T.; et al. Plasma extracellular RNA profiles in healthy and cancer patients. Sci. Rep. 2016, 6, 19413. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Chen, M.; Greening, D.W.; He, W.; Rai, A.; Zhang, W.; Simpson, R.J. Deep sequencing of RNA from three different extracellular vesicle (EV) subtypes released from the human LIM1863 colon cancer cell line uncovers distinct miRNA-enrichment signatures. PLoS ONE 2014, 9, e110314. [Google Scholar] [CrossRef] [PubMed]
- Fernando, M.R.; Jiang, C.; Krzyzanowski, G.D.; Ryan, W.L. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS ONE 2017, 12, e0183915. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, G.J.; Vallender, E.J.; Garrett, M.R.; Challagundla, L.; Overholser, J.C.; Jurjus, G.; Dieter, L.; Syed, M.; Romero, D.G.; Benghuzzi, H.; et al. Altered neuro-inflammatory gene expression in hippocampus in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 82, 177–186. [Google Scholar] [CrossRef]
- Kirk, E.; Chin, L.S.; Li, L. GRIF1 binds Hrs and is a new regulator of endosomal trafficking. J. Cell Sci. 2006, 119, 4689–4701. [Google Scholar] [CrossRef][Green Version]
- Zhang, F.; Ren, G.; Lu, Y.; Jin, B.; Wang, J.; Chen, X.; Liu, Z.; Li, K.; Nie, Y.; Wang, X.; et al. Identification of TRAK1 (Trafficking protein, kinesin-binding 1) as MGb2-Ag: A novel cancer biomarker. Cancer Lett. 2009, 274, 250–258. [Google Scholar] [CrossRef]
- Cao, Y.; Yu, S.L.; Wang, Y.; Guo, G.Y.; Ding, Q.; An, R.H. MicroRNA-dependent regulation of PTEN after arsenic trioxide treatment in bladder cancer cell line T24. Tumour Biol. 2011, 32, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Pichiorri, F.; Suh, S.S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890. [Google Scholar] [CrossRef] [PubMed]
- Hiranuma, C.; Kawakami, K.; Oyama, K.; Ota, N.; Omura, K.; Watanabe, G. Hypermethylation of the MYOD1 gene is a novel prognostic factor in patients with colorectal cancer. Int. J. Mol. Med. 2004, 13, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, A.; Muller, H.M.; Fiegl, H.; Ivarsson, L.; Wiedemair, A.; Muller-Holzner, E.; Goebel, G.; Marth, C.; Widschwendter, M. DNA methylation in serum and tumors of cervical cancer patients. Clin. Cancer Res. 2004, 10, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Hideshima, T.; Carrasco, D.; Shammas, M.; Auclair, D.; Davies, F.; Mitsiades, N.; Mitsiades, C.; Kim, R.S.; Li, C.; et al. Identification of genes modulated in multiple myeloma using genetically identical twin samples. Blood 2004, 103, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.; Park, S.; Lee, E.K.; Yoo, Y.J. Downregulation of ubiquitin level via knockdown of polyubiquitin gene Ubb as potential cancer therapeutic intervention. Sci. Rep. 2013, 3, 2623. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ren, Y.; Zuo, T. Long noncoding RNA LINC00978 promotes cell proliferation and invasion in nonsmall cell lung cancer by inhibiting miR67545p. Mol. Med. Rep. 2018, 18, 4725–4732. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.G.; Pontes, C.; Guino, E.; Navarro, M.; Osorio, A.; Canzian, F.; Moreno, V.; Bellvitge Colorectal Cancer Study, G. Polymorphisms in prostaglandin synthase 2/cyclooxygenase 2 (PTGS2/COX2) and risk of colorectal cancer. Br. J. Cancer 2004, 91, 339–343. [Google Scholar] [CrossRef]
- Langsenlehner, U.; Yazdani-Biuki, B.; Eder, T.; Renner, W.; Wascher, T.C.; Paulweber, B.; Weitzer, W.; Samonigg, H.; Krippl, P. The cyclooxygenase-2 (PTGS2) 8473T>C polymorphism is associated with breast cancer risk. Clin. Cancer Res. 2006, 12, 1392–1394. [Google Scholar] [CrossRef]
- Matsubara, J.; Honda, K.; Ono, M.; Tanaka, Y.; Kobayashi, M.; Jung, G.; Yanagisawa, K.; Sakuma, T.; Nakamori, S.; Sata, N.; et al. Reduced plasma level of CXC chemokine ligand 7 in patients with pancreatic cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 160–171. [Google Scholar] [CrossRef]
- Timms, J.F.; Menon, U.; Devetyarov, D.; Tiss, A.; Camuzeaux, S.; McCurrie, K.; Nouretdinov, I.; Burford, B.; Smith, C.; Gentry-Maharaj, A.; et al. Early detection of ovarian cancer in samples pre-diagnosis using CA125 and MALDI-MS peaks. Cancer Genom. Proteom. 2011, 8, 289–305. [Google Scholar]
- Clarke, C.H.; Yip, C.; Badgwell, D.; Fung, E.T.; Coombes, K.R.; Zhang, Z.; Lu, K.H.; Bast, R.C., Jr. Proteomic biomarkers apolipoprotein A1, truncated transthyretin and connective tissue activating protein III enhance the sensitivity of CA125 for detecting early stage epithelial ovarian cancer. Gynecol. Oncol. 2011, 122, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Yee, J.; Sadar, M.D.; Sin, D.D.; Kuzyk, M.; Xing, L.; Kondra, J.; McWilliams, A.; Man, S.F.; Lam, S. Connective tissue-activating peptide III: A novel blood biomarker for early lung cancer detection. J. Clin. Oncol. 2009, 27, 2787–2792. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.K.; Kumar, D.; Naik, M.; Dixit, M. The expression profile and prognostic significance of eukaryotic translation elongation factors in different cancers. PLoS ONE 2018, 13, e0191377. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Li, E.; Liu, Y.; Xie, W.; Huang, C.; Song, J.; Zhang, W.; Zheng, Y.; Wang, H.; Wang, Q. CTAPIII/CXCL7: A novel biomarker for early diagnosis of lung cancer. Cancer Med. 2018, 7, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Feve, M.; Saliou, J.M.; Zeniou, M.; Lennon, S.; Carapito, C.; Dong, J.; Van Dorsselaer, A.; Junier, M.P.; Chneiweiss, H.; Cianferani, S.; et al. Comparative expression study of the endo-G protein coupled receptor (GPCR) repertoire in human glioblastoma cancer stem-like cells, U87-MG cells and non malignant cells of neural origin unveils new potential therapeutic targets. PLoS ONE 2014, 9, e91519. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.P.; Barros-Silva, J.D.; Vieira, J.; Lisboa, S.; Torres, L.; Correia, C.; Vieira-Coimbra, M.; Martins, A.T.; Jeronimo, C.; Henrique, R.; et al. NCOA2 is a candidate target gene of 8q gain associated with clinically aggressive prostate cancer. Genes Chromosom. Cancer 2016, 55, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Bera, T.K.; Zimonjic, D.B.; Popescu, N.C.; Sathyanarayana, B.K.; Kumar, V.; Lee, B.; Pastan, I. POTE, a highly homologous gene family located on numerous chromosomes and expressed in prostate, ovary, testis, placenta, and prostate cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 16975–16980. [Google Scholar] [CrossRef]
- Hamidi, T.; Algul, H.; Cano, C.E.; Sandi, M.J.; Molejon, M.I.; Riemann, M.; Calvo, E.L.; Lomberk, G.; Dagorn, J.C.; Weih, F.; et al. Nuclear protein 1 promotes pancreatic cancer development and protects cells from stress by inhibiting apoptosis. J. Clin. Investig. 2012, 122, 2092–2103. [Google Scholar] [CrossRef]
- Ria, R.; Todoerti, K.; Berardi, S.; Coluccia, A.M.; De Luisi, A.; Mattioli, M.; Ronchetti, D.; Morabito, F.; Guarini, A.; Petrucci, M.T.; et al. Gene expression profiling of bone marrow endothelial cells in patients with multiple myeloma. Clin. Cancer Res. 2009, 15, 5369–5378. [Google Scholar] [CrossRef]
- Samur, M.K.; Minvielle, S.; Gulla, A.; Fulciniti, M.; Cleynen, A.; Aktas Samur, A.; Szalat, R.; Shammas, M.; Magrangeas, F.; Tai, Y.T.; et al. Long intergenic non-coding RNAs have an independent impact on survival in multiple myeloma. Leukemia 2018, 32, 2626–2635. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, D.; Agnelli, L.; Pietrelli, A.; Todoerti, K.; Manzoni, M.; Taiana, E.; Neri, A. A compendium of long non-coding RNAs transcriptional fingerprint in multiple myeloma. Sci. Rep. 2018, 8, 6557. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, L.; Dasoula, A.; Hatzimichael, E.; Georgiou, I.; Syrrou, M.; Bourantas, K.L. Promoter hypermethylation of the MEG3 (DLK1/MEG3) imprinted gene in multiple myeloma. Clin. Lymphoma Myeloma 2008, 8, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.F.; Chang, Y.C.; Chang, C.S.; Lin, S.F.; Liu, Y.C.; Hsiao, H.H.; Chang, J.G.; Liu, T.C. MALAT1 long non-coding RNA is overexpressed in multiple myeloma and may serve as a marker to predict disease progression. BMC Cancer 2014, 14, 809. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.B.; He, X.; Huang, Y.F.; Wu, Q.N.; Zhou, Y.C.; Hao, D.J. Long Noncoding RNA CRNDE Promotes Multiple Myeloma Cell Growth by Suppressing miR-451. Oncol. Res. 2017, 25, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, H.; Shen, X.; Wang, X.; Ju, S.; Lu, M.; Cong, H. Serum level of long noncoding RNA H19 as a diagnostic biomarker of multiple myeloma. Clin. Chim. Acta 2018, 480, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.B.; Dong, C.P.; Zhou, G.C.; Lu, S.M.; Luan, Y.; Gu, X.; Liu, L.; Ding, X.F. A potential panel of four-long noncoding RNA signature in prostate cancer predicts biochemical recurrence-free survival and disease-free survival. Int. Urol. Nephrol. 2017, 49, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Grasedieck, S.; Rueß, C.; Pochert, N.; Lux, S.; Schwarzer, A.; Klusmann, J.-H.; Jongen-Lavrencic, M.; Pollack, J.R.; Rouhi, A.; Kuchenbauer, F. Identification of Novel Lncrnas That Predict Survival in AML Patients and Modulate Leukemic Cells. Blood 2018, 132, 3909. [Google Scholar] [CrossRef]
- Zhou, B.; Guo, W.; Sun, C.; Zhang, B.; Zheng, F. Linc00462 promotes pancreatic cancer invasiveness through the miR-665/TGFBR1-TGFBR2/SMAD2/3 pathway. Cell Death Dis. 2018, 9, 706. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Morley, R.; Khong, T.; Kalff, A.; Bergin, K.; Hocking, J.; Savvidou, I.; Bowen, K.M.; Ramachandran, M.; Choi, K.; et al. Monitoring tumour burden and therapeutic response through analysis of circulating tumour DNA and extracellular RNA in multiple myeloma patients. Leukemia 2019. [Google Scholar] [CrossRef]
- Piskol, R.; Ramaswami, G.; Li, J.B. Reliable identification of genomic variants from RNA-seq data. Am. J. Hum. Genet. 2013, 93, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pasche, B.; Zhang, W.; Chen, K. Association of MUC16 Mutation with Tumor Mutation Load and Outcomes in Patients with Gastric Cancer. JAMA Oncol. 2018, 4, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- King, R.J.; Yu, F.; Singh, P.K. Genomic alterations in mucins across cancers. Oncotarget 2017, 8, 67152–67168. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).