The 6th R of Radiobiology: Reactivation of Anti-Tumor Immune Response

 ,
, 
Abstract
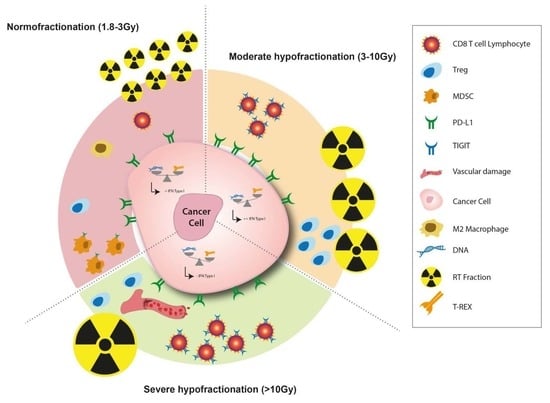
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Radiation Therapy Effects: From DNA Breaks to Immunomodulation
- Repair: the capacity to repair radiation-induced DNA damage.
- Re-assortment: cells in mitosis are mostly sensitive to radiation, whereas cells in S-phase are mostly resistant. With multiple doses, cells progress through to a sensitive phase of the cell cycle, which leads to the therapeutic effect.
- Repopulation: cell proliferation between fractions.
- Reoxygenation: the sensitivity to radiation increases in well oxygenated tissues.
- Radiosensitivity: the response to radiation varies by tumor intrinsic and individual radiosensitivity.
2. Overcoming Radiation-Induced Immune Resistance
2.1. Modulation of Radiation Therapy
2.1.1. Finding the Optimal RT Dose and Fractionation to Induce Anti-Tumor Immune Response
2.1.2. The Immunoregulatory Potential of Particle Radiation: Hadrontherapy versus Proton Therapy
2.2. Combination of RT and Immune Checkpoint Inhibitors
2.2.1. Anti-CTLA-4, PD-1 and PD-L1
2.2.2. Other Immunotherapies
2.3. Optimization of RT-Immunotherapy Combination
3. Prospects About Associating Focal Irradiation with Biotherapies Targeting Recently Discovered Immune Pathways: Toward a Promising New Weapon in the Anti-Cancer Armamentarium?
- Simultaneous research on the optimal fractionation and best RT modalities to efficiently promote the action of a concomitantly-administered drug targeting immune checkpoints. These conditions should differ depending on the drug that is used, and careful reevaluation should be done for each novel molecule.
- Integrating, in early-phase clinical trials investigating novel immune-checkpoint blockers under development in pharmaceutical pipelines, their association to RT, before their approval by medical drugs agencies such as the Food and Drug Administration (FDA) in the United States or the European Medicines Agency (EMA) in Europe. This should contribute to their optimal use once marketed.
- The development of clinical and biological tools that can be used routinely to help clinicians identify the patients most likely to benefit from a therapeutic scheme combining new ICI and RT and to help them decide on the most appropriate RT schemes to be used.
3.1. Association of Specific Chemotherapy with RT and Immunotherapy
3.2. Association of Extreme Hypofractionated RT with STING Agonist
3.3. TIM-3 and TIGIT: A Contemporary Lack of Data Leading Toward a Promising Future?
3.4. Combination Treatments Associating RT with ICI-Based Immunotherapy: What If Personalization Was the Keyword?
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gao, D. Compound-therapy based on cancer-immunity cycle: Promising prospects for antitumor regimens. Am. J. Cancer Res. 2019, 9, 212–218. [Google Scholar] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Withers, H.R. The Four R’s of radiotherapy. Adv. Radiat. Biol. 1975, 5, 241–247. [Google Scholar]
- Steel, G.G.; McMillan, T.J.; Peacock, J.H. The 5Rs of Radiobiology. Int. J. Radiat. Biol. 1989, 56, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Young, A.; Berry, R.; Holloway, A.F.; Blackburn, N.B.; Dickinson, J.L.; Skala, M.; Phillips, J.L.; Brettingham-Moore, K.H. RNA-seq profiling of a radiation resistant and radiation sensitive prostate cancer cell line highlights opposing regulation of DNA repair and targets for radiosensitization. BMC Cancer 2014, 14, 808. [Google Scholar] [CrossRef]
- Purbey, P.K.; Scumpia, P.O.; Kim, P.J.; Tong, A.-J.; Iwamoto, K.S.; McBride, W.H.; Smale, S.T. Defined Sensing Mechanisms and Signaling Pathways Contribute to the Global Inflammatory Gene Expression Output Elicited by Ionizing Radiation. Immunity 2017, 47, 421–434.e3. [Google Scholar] [CrossRef]
- Stone, H.B.; Peters, L.J.; Milas, L. Effect of host immune capability on radiocurability and subsequent transplantability of a murine fibrosarcoma. J. Natl. Cancer Inst. 1979, 63, 1229–1235. [Google Scholar] [PubMed]
- Turgeon, G.-A.; Weickhardt, A.; Azad, A.A.; Solomon, B.; Siva, S. Radiotherapy and immunotherapy: A synergistic effect in cancer care. Med. J. Aust. 2019, 210, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Sato, H.; Multhoff, G.; Shibata, A. Novel Approaches to Improve the Efficacy of Immuno-Radiotherapy. Front. Oncol. 2019, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef]
- Hallahan, D.; Kuchibhotla, J.; Wyble, C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res. 1996, 56, 5150–5155. [Google Scholar] [PubMed]
- Ishihara, H.; Tsuneoka, K.; Dimchev, A.B.; Shikita, M. Induction of the expression of the interleukin-1 beta gene in mouse spleen by ionizing radiation. Radiat. Res. 1993, 133, 321–326. [Google Scholar] [CrossRef]
- Hallahan, D.E.; Spriggs, D.R.; Beckett, M.A.; Kufe, D.W.; Weichselbaum, R.R. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc. Natl. Acad. Sci. USA 1989, 86, 10104–10107. [Google Scholar] [CrossRef]
- Lugade, A.A.; Sorensen, E.W.; Gerber, S.A.; Moran, J.P.; Frelinger, J.G.; Lord, E.M. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J. Immunol. 2008, 180, 3132–3139. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Garnett, C.T.; Palena, C.; Chakraborty, M.; Chakarborty, M.; Tsang, K.-Y.; Schlom, J.; Hodge, J.W. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004, 64, 7985–7994. [Google Scholar] [CrossRef] [PubMed]
- Vereecque, R.; Buffenoir, G.; Gonzalez, R.; Cambier, N.; Hetuin, D.; Bauters, F.; Fenaux, P.; Quesnel, B. gamma-ray irradiation induces B7.1 expression in myeloid leukaemic cells. Br. J. Haematol. 2000, 108, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Son, Y.-O.; Park, S.-W.; Bae, J.-H.; Chung, J.S.; Kim, H.H.; Chung, B.-S.; Kim, S.-H.; Kang, C.-D. Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp. Mol. Med. 2006, 38, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef]
- Yilmaz, M.T.; Elmali, A.; Yazici, G. Abscopal Effect, From Myth to Reality: From Radiation Oncologists’ Perspective. Cureus 2019, 11, e3860. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-S.; Chen, F.-H.; Wang, C.-C.; Huang, H.-L.; Jung, S.-M.; Wu, C.-J.; Lee, C.-C.; McBride, W.H.; Chiang, C.-S.; Hong, J.-H. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 499–507. [Google Scholar] [CrossRef]
- Chiang, C.-S.; Fu, S.Y.; Wang, S.-C.; Yu, C.-F.; Chen, F.-H.; Lin, C.-M.; Hong, J.-H. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front. Oncol. 2012, 2, 89. [Google Scholar] [CrossRef]
- Schaue, D.; Xie, M.W.; Ratikan, J.A.; McBride, W.H. Regulatory T cells in radiotherapeutic responses. Front. Oncol. 2012, 2, 90. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, S.E.; Beiske, K.; Blaker, Y.N.; Førsund, M.S.; Holte, H.; Østenstad, B.; Kimby, E.; Köksal, H.; Wälchli, S.; Bai, B.; et al. TIGIT and PD-1 Mark Intratumoral T Cells with Reduced Effector Function in B-cell Non-Hodgkin Lymphoma. Cancer Immunol. Res. 2019, 7, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Grapin, M.; Limagne, E.; Richard, C.; Boidot, R.; Morgand, V.; Bertaut, A.; Derangere, V.; Laurent, P.A.; Thibaudin, M.; Fumet, J.D.; et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: A promising new combination. J ImmunoTher. Cancer. 2019. [Google Scholar] [CrossRef]
- Casado, J.G.; Pawelec, G.; Morgado, S.; Sanchez-Correa, B.; Delgado, E.; Gayoso, I.; Duran, E.; Solana, R.; Tarazona, R. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol. Immunother. 2009, 58, 1517–1526. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhu, L.; Schell, T.D.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; George, M.R.; Zeng, H.; Zheng, H. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin. Cancer Res. 2016, 22, 3057–3066. [Google Scholar] [CrossRef] [PubMed]
- Yovino, S.; Kleinberg, L.; Grossman, S.A.; Narayanan, M.; Ford, E. The Etiology of Treatment-related Lymphopenia in Patients with Malignant Gliomas: Modeling Radiation Dose to Circulating Lymphocytes Explains Clinical Observations and Suggests Methods of Modifying the Impact of Radiation on Immune Cells. Cancer Investig. 2013, 31, 140–144. [Google Scholar] [CrossRef]
- Serre, R.; Barlesi, F.; Muracciole, X.; Barbolosi, D. Immunologically effective dose: A practical model for immuno-radiotherapy. Oncotarget 2018, 9, 31812–31819. [Google Scholar] [CrossRef][Green Version]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Rivera Vargas, T.; Apetoh, L. Danger signals: Chemotherapy enhancers? Immunol. Rev. 2017, 280, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Griffin, R.J.; Hui, S.; Levitt, S.H.; Song, C.W. Radiation-Induced Vascular Damage in Tumors: Implications of Vascular Damage in Ablative Hypofractionated Radiotherapy (SBRT and SRS). Radiat. Res. 2012, 177, 311–327. [Google Scholar] [CrossRef]
- Poleszczuk, J.; Enderling, H. The Optimal Radiation Dose to Induce Robust Systemic Anti-Tumor Immunity. Int. J. Mol. Sci. 2018, 19, 3377. [Google Scholar] [CrossRef]
- Wild, A.T.; Herman, J.M.; Dholakia, A.S.; Moningi, S.; Lu, Y.; Rosati, L.M.; Hacker-Prietz, A.; Assadi, R.K.; Saeed, A.M.; Pawlik, T.M.; et al. Lymphocyte-Sparing Effect of Stereotactic Body Radiation Therapy in Patients with Unresectable Pancreatic Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 571–579. [Google Scholar] [CrossRef]
- Wunderlich, R.; Ernst, A.; Rödel, F.; Fietkau, R.; Ott, O.; Lauber, K.; Frey, B.; Gaipl, U.S. Low and moderate doses of ionizing radiation up to 2 Gy modulate transmigration and chemotaxis of activated macrophages, provoke an anti-inflammatory cytokine milieu, but do not impact upon viability and phagocytic function. Clin. Exp. Immunol. 2015, 179, 50–61. [Google Scholar] [CrossRef]
- Tsai, M.-H.; Cook, J.A.; Chandramouli, G.V.R.; DeGraff, W.; Yan, H.; Zhao, S.; Coleman, C.N.; Mitchell, J.B.; Chuang, E.Y. Gene expression profiling of breast, prostate, and glioma cells following single versus fractionated doses of radiation. Cancer Res. 2007, 67, 3845–3852. [Google Scholar] [CrossRef]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef]
- Prakash, H.; Klug, F.; Nadella, V.; Mazumdar, V.; Schmitz-Winnenthal, H.; Umansky, L. Low doses of gamma irradiation potentially modifies immunosuppressive tumor microenvironment by retuning tumor-associated macrophages: Lesson from insulinoma. Carcinogenesis 2016, 37, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Nguy, S.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Daley, D.; Barilla, R.; et al. Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology 2016, 150, 1659–1672.e5. [Google Scholar] [CrossRef] [PubMed]
- Kachikwu, E.L.; Iwamoto, K.S.; Liao, Y.-P.; DeMarco, J.J.; Agazaryan, N.; Economou, J.S.; McBride, W.H.; Schaue, D. Radiation enhances regulatory T cell representation. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Qinfeng, S.; Depu, W.; Xiaofeng, Y.; Shah, W.; Hongwei, C.; Yili, W. In situ observation of the effects of local irradiation on cytotoxic and regulatory T lymphocytes in cervical cancer tissue. Radiat. Res. 2013, 179, 584–589. [Google Scholar] [CrossRef]
- Schaue, D.; Ratikan, J.A.; Iwamoto, K.S.; McBride, W.H. Maximizing Tumor Immunity with Fractionated Radiation. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1306–1310. [Google Scholar] [CrossRef]
- Wei, S.; Egenti, M.U.; Teitz-Tennenbaum, S.; Zou, W.; Chang, A.E. Effects of tumor irradiation on host T-regulatory cells and systemic immunity in the context of adoptive T-cell therapy in mice. J. Immunother. 2013, 36, 124–132. [Google Scholar] [CrossRef]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef]
- Lan, J.; Li, R.; Yin, L.-M.; Deng, L.; Gui, J.; Chen, B.-Q.; Zhou, L.; Meng, M.-B.; Huang, Q.-R.; Mo, X.-M.; et al. Targeting Myeloid-derived Suppressor Cells and Programmed Death Ligand 1 Confers Therapeutic Advantage of Ablative Hypofractionated Radiation Therapy Compared with Conventional Fractionated Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 74–87. [Google Scholar] [CrossRef]
- Crittenden, M.R.; Savage, T.; Cottam, B.; Bahjat, K.S.; Redmond, W.L.; Bambina, S.; Kasiewicz, M.; Newell, P.; Jackson, A.M.; Gough, M.J. The peripheral myeloid expansion driven by murine cancer progression is reversed by radiation therapy of the tumor. PLoS ONE 2013, 8, e69527. [Google Scholar] [CrossRef]
- Arnold, K.M.; Flynn, N.J.; Raben, A.; Romak, L.; Yu, Y.; Dicker, A.P.; Mourtada, F.; Sims-Mourtada, J. The Impact of Radiation on the Tumor Microenvironment: Effect of Dose and Fractionation Schedules. Cancer Growth Metastasis 2018, 11. [Google Scholar] [CrossRef]
- Rödel, F.; Frey, B.; Manda, K.; Hildebrandt, G.; Hehlgans, S.; Keilholz, L.; Seegenschmiedt, M.H.; Gaipl, U.S.; Rödel, C. Immunomodulatory Properties and Molecular Effects in Inflammatory Diseases of Low-Dose X-Irradiation. Front. Oncol. 2012, 2, 120. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.; Brenner, D.J.; Formenti, S.C. Does Heavy Ion Therapy Work Through the Immune System? Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 934–936. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.; Orecchia, R.; Loeffler, J.S. Charged-particle therapy in cancer: Clinical uses and future perspectives. Nat. Rev. Clin. Oncol. 2017, 14, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lupu-Plesu, M.; Claren, A.; Martial, S.; N’Diaye, P.-D.; Lebrigand, K.; Pons, N.; Ambrosetti, D.; Peyrottes, I.; Feuillade, J.; Hérault, J.; et al. Effects of proton versus photon irradiation on (lymph)angiogenic, inflammatory, proliferative and anti-tumor immune responses in head and neck squamous cell carcinoma. Oncogenesis 2017, 6, e354. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Demizu, Y.; Hashimoto, N.; Mima, M.; Terashima, K.; Fujii, O.; Jin, D.; Niwa, Y.; Morimoto, K.; Akagi, T.; et al. Treatment outcomes of particle radiotherapy using protons or carbon ions as a single-modality therapy for adenoid cystic carcinoma of the head and neck. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2014, 113, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Shin, S.; Dy, G. Advances in Cancer Immunotherapy in Solid Tumors. Cancers 2016, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Smyth, M.J.; Drake, C.G.; Abastado, J.-P.; Apte, R.N.; Ayyoub, M.; Blay, J.-Y.; Bonneville, M.; Butterfield, L.H.; Caignard, A.; et al. Consensus nomenclature for CD8+ T cell phenotypes in cancer. Oncoimmunology 2015, 4, e998538. [Google Scholar] [CrossRef]
- Demaria, S.; Pilones, K.A.; Vanpouille-Box, C.; Golden, E.B.; Formenti, S.C. The optimal partnership of radiation and immunotherapy: From preclinical studies to clinical translation. Radiat. Res. 2014, 182, 170–181. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.M.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef]
- Ribeiro Gomes, J.; Schmerling, R.A.; Haddad, C.K.; Racy, D.J.; Ferrigno, R.; Gil, E.; Zanuncio, P.; Buzaid, A.C. Analysis of the Abscopal Effect with Anti-PD1 Therapy in Patients with Metastatic Solid Tumors. J. Immunother. 2016, 39, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F.; et al. Focal Irradiation and Systemic TGFβ Blockade in Metastatic Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F. TIM-3, a promising target for cancer immunotherapy. OncoTargets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed]
- Oweida, A.; Hararah, M.K.; Phan, A.; Binder, D.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Court, B.V.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed]
- Shaverdian, N.; Lisberg, A.E.; Bornazyan, K.; Veruttipong, D.; Goldman, J.W.; Formenti, S.C.; Garon, E.B.; Lee, P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: A secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017, 18, 895–903. [Google Scholar] [CrossRef]
- Young, K.H.; Baird, J.R.; Savage, T.; Cottam, B.; Friedman, D.; Bambina, S.; Messenheimer, D.J.; Fox, B.; Newell, P.; Bahjat, K.S.; et al. Optimizing Timing of Immunotherapy Improves Control of Tumors by Hypofractionated Radiation Therapy. PLoS ONE 2016, 11, e0157164. [Google Scholar] [CrossRef]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Evans, D.E.; Prell, R.A.; Thalhofer, C.J.; Hurwitz, A.A.; Weinberg, A.D. Engagement of OX40 enhances antigen-specific CD4(+) T cell mobilization/memory development and humoral immunity: Comparison of alphaOX-40 with alphaCTLA-4. J. Immunol. 2001, 167, 6804–6811. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rebe, C.; Ghiringhelli, F. 5-Fluorouracil Selectively Kills Tumor-Associated Myeloid-Derived Suppressor Cells Resulting in Enhanced T Cell-Dependent Antitumor Immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef]
- Brode, S.; Cooke, A. Immune-potentiating effects of the chemotherapeutic drug cyclophosphamide. Crit. Rev. Immunol. 2008, 28, 109–126. [Google Scholar] [CrossRef]
- Preliminary Results of the First-in-Human (FIH) Study of MK-1454, an Agonist of Stimulator of Interferon Genes (STING), as Monotherapy or in Combin… | OncologyPRO. Available online: https://oncologypro.esmo.org/Meeting-Resources/ESMO-2018-Congress/Preliminary-results-of-the-first-in-human-FIH-study-of-MK-1454-an-agonist-of-stimulator-of-interferon-genes-STING-as-monotherapy-or-in-combination-with-pembrolizumab-pembro-in-patients-with-advanced-solid-tumors-or-lymphomas (accessed on 24 February 2019).
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.-P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.L.; Garrido-Laguna, I. TIGIT: A novel immunotherapy target moving from bench to bedside. Cancer Immunol. Immunother. 2018, 67, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.-F.; Joller, N.; Tan, D.J.; Teng, M.W.L.; Smyth, M.J.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Investig. 2015, 125, 4053–4062. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. OncoImmunology 2018, 7, e1466769. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Formenti, S.C.; Demaria, S. Toward Precision Radiotherapy for Use with Immune Checkpoint Blockers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 259–265. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boustani, J.; Grapin, M.; Laurent, P.-A.; Apetoh, L.; Mirjolet, C. The 6th R of Radiobiology: Reactivation of Anti-Tumor Immune Response. Cancers 2019, 11, 860. https://doi.org/10.3390/cancers11060860
Boustani J, Grapin M, Laurent P-A, Apetoh L, Mirjolet C. The 6th R of Radiobiology: Reactivation of Anti-Tumor Immune Response. Cancers. 2019; 11(6):860. https://doi.org/10.3390/cancers11060860
Chicago/Turabian StyleBoustani, Jihane, Mathieu Grapin, Pierre-Antoine Laurent, Lionel Apetoh, and Céline Mirjolet. 2019. "The 6th R of Radiobiology: Reactivation of Anti-Tumor Immune Response" Cancers 11, no. 6: 860. https://doi.org/10.3390/cancers11060860
APA StyleBoustani, J., Grapin, M., Laurent, P.-A., Apetoh, L., & Mirjolet, C. (2019). The 6th R of Radiobiology: Reactivation of Anti-Tumor Immune Response. Cancers, 11(6), 860. https://doi.org/10.3390/cancers11060860