Gold as a Possible Alternative to Platinum-Based Chemotherapy for Colon Cancer Treatment




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Gold(I) Complexes
2.1. Phosphane Gold(I) Derivatives
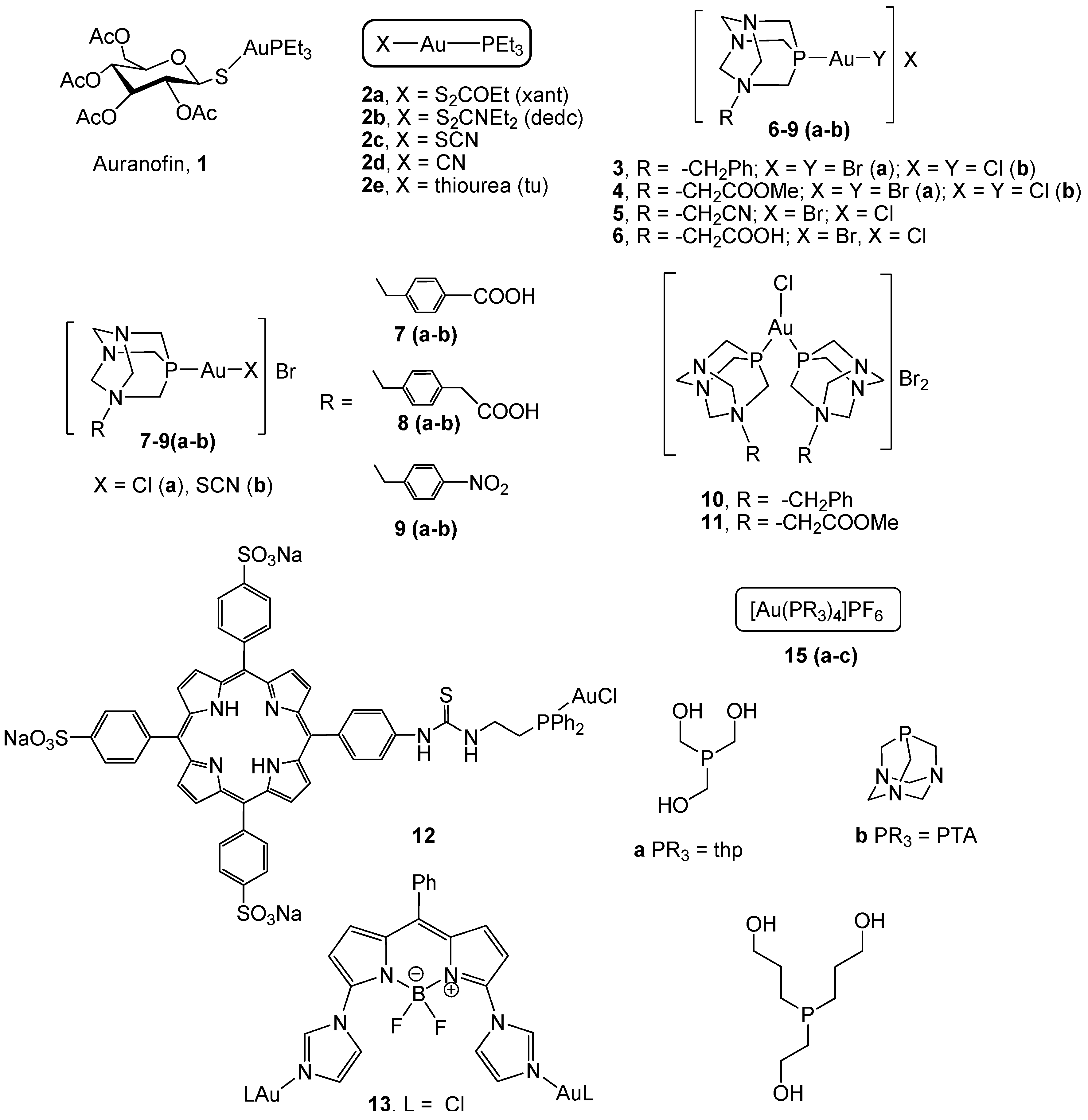
2.2. Gold(I) Complexes With S-Donor Ligands
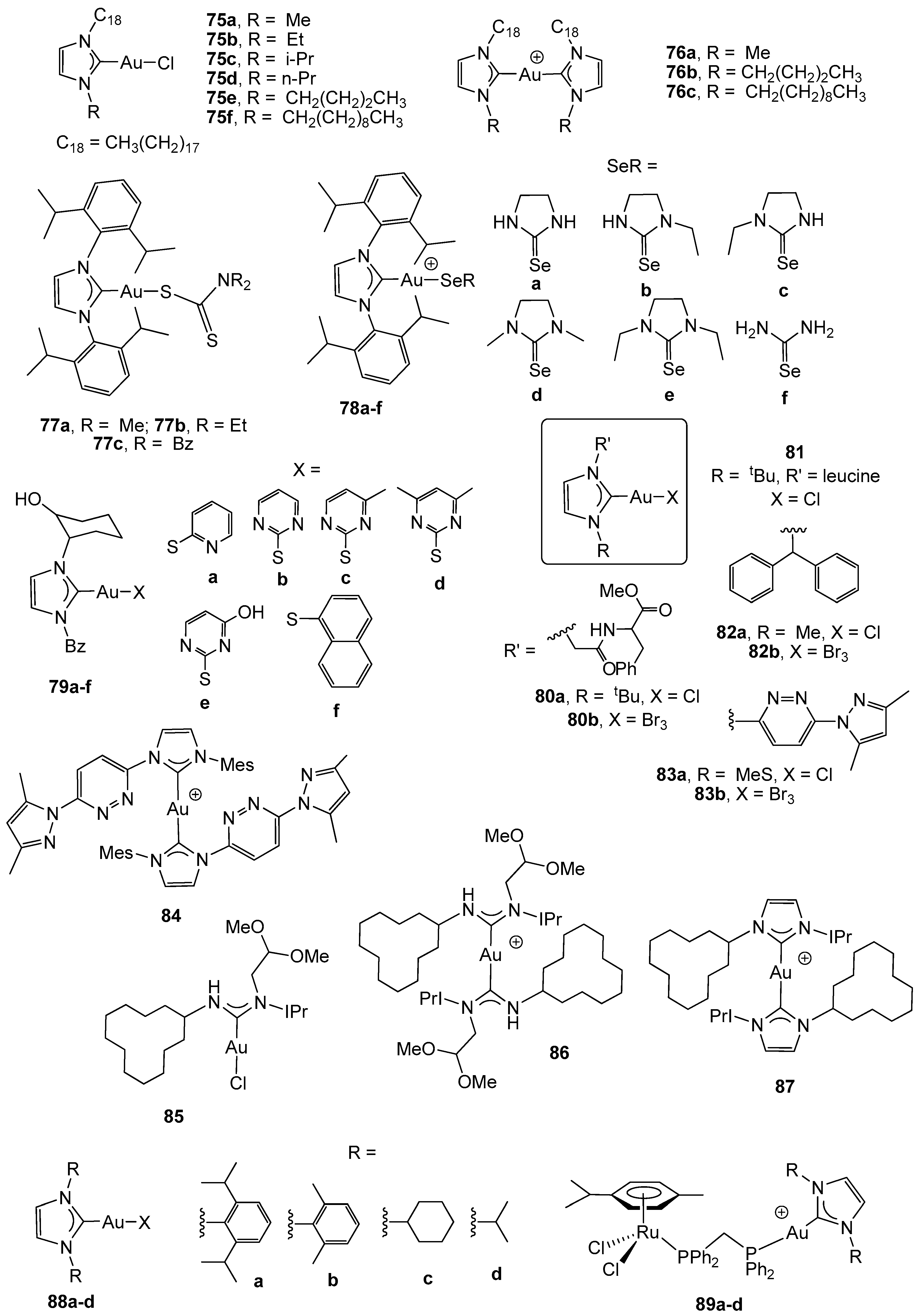
2.3. Carbene Gold(I) Derivatives
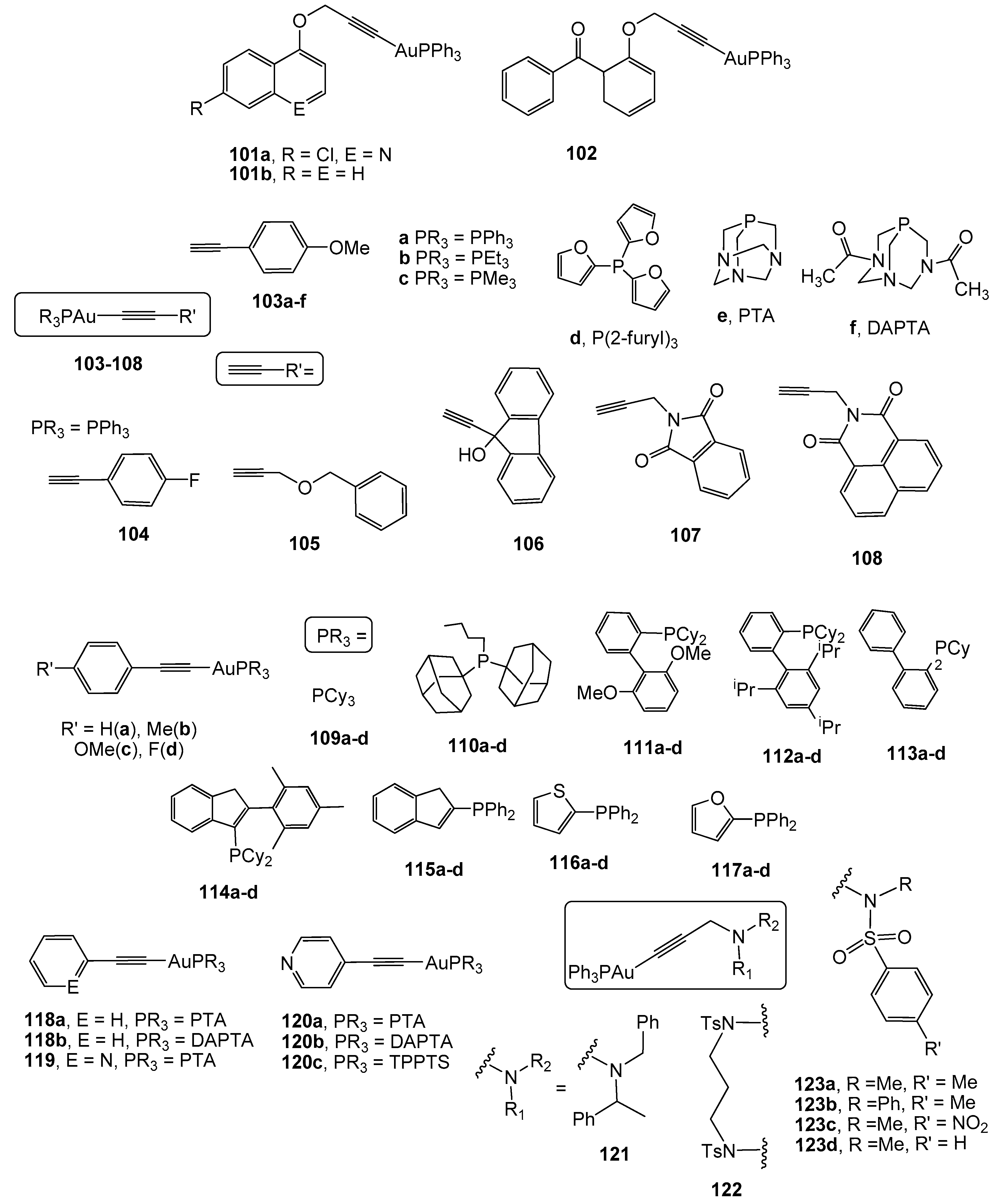
2.4. Alkynyl Gold(I) Derivatives
3. Gold as a Vehicle: Biomedical Potential of Gold Nanoparticles in CRC Therapy
3.1. Basis of Surface Plasmon Resonance in GNPs
3.2. GNPs as Contrast Agents
3.3. Gold Nanoparticles as Photothermal Agents
3.4. Gold Nanoparticles in Photodynamic Therapy
3.5. Gold Nanoparticles as Radiosensitizers
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PP | 1,2-bis-(diphenylphosphino)ethane |
| PTA | 1,3,5-triaza-7-phosphaadamantane |
| IPr | 1,3-Bis(2,6-diisopropylphenyl) imidazol-2-ylidene |
| dppb | 1,4-bis(diphenylphosphino)butane |
| DAPTA | 3,7-diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane |
| BCL-2 | B-cell CLL/lymphoma 2 |
| dppm | Bis(diphenylphosphino)methane |
| CA | Carbonic anhydrase |
| CRC | Colorectal cancer |
| CT | Computed tomography |
| COX | Cyclooxygenase |
| Cys | Cysteine |
| DLC | Delocalised lipophilic cations |
| dppp | Diphosphane 1,3-bis(diphenylphosphino)propane |
| DDS | Drug delivery system |
| EPR | Enhanced permeability and retention |
| EGFR | Epidermal growth factor receptor |
| GSH | Gluthathione |
| GSSG | Glutathione disulphide |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GNP | Gold nanoparticle |
| iNOS | Inducible nitric oxide synthase |
| MRI | Magnetic resonance imaging |
| MAPK | Mitogen activated kinases |
| NAC | N-acyclic carbene |
| NHC | N-heterocyclic carbene |
| NIR | Near-infrared |
| NO | Nitric oxide |
| OCT | Optical coherence tomography |
| PAM | Photoacoustic microscopy |
| PDT | Photodynamic therapy |
| PTT | Photothermal therapy |
| PARP-1 | Poly(ADP-ribose) polymerase 1 |
| PEI | Polyethyleneniumine |
| PEG | Polyethylene glycol |
| PLGA | Poly(lactic-co-glicolic acid) |
| PD-L1 | Programmed death-ligand 1 |
| RT | Radiotherapy |
| ROS | Reactive oxygen species |
| Sec | Selenocysteine |
| SAR | Structure-activity relationship |
| SPR | Surface plasmon resonance |
| Trx | Thioredoxin |
| TrxR | Thioredoxin reductase |
| thp | Tris(hydroxymethyl)phosphine |
| thpp | Tris(hydroxypropyl)phosphine |
| TPPTS | Triphenylphosphane trisulphonated |
| TNF | Tumor necrosis factor |
| TNFR | Tumor necrosis factor receptor |
| (UPS) | Ubiquitin/proteasome system |
| VEGF | Vascular endothelial growth factor |
| C11Pc | Zinc phthalocyanine disulphide |
References
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Marmol, I.; Sanchez-De-Diego, C.; Dieste, A.P.; Cerrada, E.; Yoldi, M.J.R. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Mármol, I.; Quero, J.; Rodriguez Yoldi, M.J. Modifiable and non-modifiable risk factors of colorectal cancer. In Advances in Health and Disease, 1st ed.; Duncan, L.T., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2018; Volume 7, pp. 67–116. [Google Scholar]
- Lee, M.M.; MacKinlay, A.; Semira, C.; Schieber, C.; Jimeno Yepes, A.J.; Lee, B.; Wong, R.; Hettiarachchige, C.K.H.; Gunn, N.; Tie, J.; et al. Stage-based Variation in the Effect of Primary Tumor Side on All Stages of Colorectal Cancer Recurrence and Survival. Clin. Colorectal Cancer 2018, 17, e569–e577. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Wu, C.; Goldberg, R.M. Recent therapeutic advances in the treatment of colorectal cancer. Annu. Rev. Med. 2015, 66, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.M.; Cosby, R.; Quereshy, F.; Jonker, D. Adjuvant Chemotherapy for Stage II and III Colon Cancer Following Complete Resection: A Cancer Care Ontario Systematic Review. Clin. Oncol. 2017, 29, 459–465. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Misset, J.L.; Bleiberg, H.; Sutherland, W.; Bekradda, M.; Cvitkovic, E. Oxaliplatin clinical activity: A review. Crit. Rev. Oncol. Hematol. 2000, 35, 75–93. [Google Scholar] [CrossRef]
- Faivre, S.; Chan, D.; Salinas, R.; Woynarowska, B.; Woynarowski, J.M. DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells. Biochem. Pharmacol. 2003, 66, 225–237. [Google Scholar] [CrossRef]
- Bergamo, A.; Dyson, P.J.; Sava, G. The mechanism of tumour cell death by metal-based anticancer drugs is not only a matter of DNA interactions. Coord. Chem. Rev. 2018, 360, 17–33. [Google Scholar] [CrossRef]
- Palermo, G.; Magistrato, A.; Riedel, T.; von Erlach, T.; Davey, C.A.; Dyson, P.J.; Rothlisberger, U. Fighting Cancer with Transition Metal Complexes: From Naked DNA to Protein and Chromatin Targeting Strategies. Chem. Med. Chem. 2016, 11, 1199–1210. [Google Scholar] [CrossRef]
- Yiu, A.J.; Yiu, C.Y. Biomarkers in Colorectal Cancer. Anticancer Res. 2016, 36, 1093–1102. [Google Scholar] [PubMed]
- Raffel, J.; Bhattacharyya, A.K.; Gallegos, A.; Cui, H.; Einspahr, J.G.; Alberts, D.S.; Powis, G. Increased expression of thioredoxin-1 in human colorectal cancer is associated with decreased patient survival. J. Lab. Clin. Med. 2003, 142, 46–51. [Google Scholar] [CrossRef]
- Arner, E.S.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef]
- Karlenius, T.C.; Tonissen, K.F. Thioredoxin and Cancer: A Role for Thioredoxin in all States of Tumor Oxygenation. Cancers 2010, 2, 209–232. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Madura, K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res. 2005, 65, 5599–5606. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Bauer, I.; Schafmayer, C.; Tepel, J.; Muerkoster, S.S.; Brosch, M.; Roder, C.; Kalthoff, H.; Hampe, J.; Moyer, M.P.; et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene 2009, 28, 3983–3996. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. The ubiquitin-proteasome system in colorectal cancer. Biochim. Biophys. Acta 2008, 1782, 800–808. [Google Scholar] [CrossRef]
- Almond, J.B.; Cohen, G.M. The proteasome: A novel target for cancer chemotherapy. Leukemia 2002, 16, 433–443. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, H.; Hu, M.; Zhang, Y.; Chen, S.; Tighe, S.; Zhu, Y. The Role of Cyclooxygenase-2 in Colorectal Carcinogenesis. Clin. Colorectal Cancer 2017, 16, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, S.; Lin, Z.; Jiang, Y.; Chen, Y.; Chen, Z.S.; Yang, X.; Gao, W. A personalized and long-acting local therapeutic platform combining photothermal therapy and chemotherapy for the treatment of multidrug-resistant colon tumor. Int. J. Nanomed. 2018, 13, 8411–8427. [Google Scholar] [CrossRef] [PubMed]
- Arisan, E.D.; Ergul, Z.; Bozdag, G.; Rencuzogullari, O.; Coker-Gurkan, A.; Obakan-Yerlikaya, P.; Coskun, D.; Palavan-Unsal, N. Diclofenac induced apoptosis via altering PI3K/Akt/MAPK signaling axis in HCT 116 more efficiently compared to SW480 colon cancer cells. Mol. Biol. Rep. 2018, 45, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Kivela, A.; Parkkila, S.; Saarnio, J.; Karttunen, T.J.; Kivela, J.; Parkkila, A.K.; Waheed, A.; Sly, W.S.; Grubb, J.H.; Shah, G.; et al. Expression of a novel transmembrane carbonic anhydrase isozyme XII in normal human gut and colorectal tumors. Am. J. Pathol. 2000, 156, 577–584. [Google Scholar] [CrossRef]
- Zhang, J.; Tsoi, H.; Li, X.; Wang, H.; Gao, J.; Wang, K.; Go, M.Y.; Ng, S.C.; Chan, F.K.; Sung, J.J.; et al. Carbonic anhydrase IV inhibits colon cancer development by inhibiting the Wnt signalling pathway through targeting the WTAP-WT1-TBL1 axis. Gut 2016, 65, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Tuluce, Y.; Ahmed, B.A.; Koyuncu, I.; Durgun, M. The cytotoxic, apoptotic and oxidative effects of carbonic anhydrase IX inhibitor on colorectal cancer cells. J. Bioenerg. Biomembr. 2018, 50, 107–116. [Google Scholar] [CrossRef]
- Dogne, J.M.; Thiry, A.; Pratico, D.; Masereel, B.; Supuran, C.T. Dual carbonic anhydrase--cyclooxygenase-2 inhibitors. Curr. Top. Med. Chem. 2007, 7, 885–891. [Google Scholar] [CrossRef]
- Vahora, H.; Khan, M.A.; Alalami, U.; Hussain, A. The Potential Role of Nitric Oxide in Halting Cancer Progression Through Chemoprevention. J. Cancer Prev. 2016, 21, 1–12. [Google Scholar] [CrossRef]
- Olah, G.; Modis, K.; Toro, G.; Hellmich, M.R.; Szczesny, B.; Szabo, C. Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation. Biochem. Pharmacol. 2018, 149, 186–204. [Google Scholar] [CrossRef]
- Chen, L.D.; Liu, Z.H.; Zhang, L.F.; Yao, J.N.; Wang, C.F. Sanggenon C induces apoptosis of colon cancer cells via inhibition of NO production, iNOS expression and ROS activation of the mitochondrial pathway. Oncol. Rep. 2017, 38, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Rim, Y.; McPherson, D.D.; Huang, S.L.; Kim, H. A novel liposomal nanomedicine for nitric oxide delivery and breast cancer treatment. Biomed. Mater. Eng. 2014, 24, 61–67. [Google Scholar] [PubMed]
- Chung, P.; Cook, T.; Liu, K.; Vodovotz, Y.; Zamora, R.; Finkelstein, S.; Billiar, T.; Blumberg, D. Overexpression of the human inducible nitric oxide synthase gene enhances radiation-induced apoptosis in colorectal cancer cells via a caspase-dependent mechanism. Nitric Oxide 2003, 8, 119–126. [Google Scholar] [CrossRef]
- Troiani, T.; Napolitano, S.; Della Corte, C.M.; Martini, G.; Martinelli, E.; Morgillo, F.; Ciardiello, F. Therapeutic value of EGFR inhibition in CRC and NSCLC: 15 years of clinical evidence. ESMO Open 2016, 1, e000088. [Google Scholar] [CrossRef] [PubMed]
- Seeber, A.; Gunsilius, E.; Gastl, G.; Pircher, A. Anti-Angiogenics: Their Value in Colorectal Cancer Therapy. Oncol. Res. Treat. 2018, 41, 188–193. [Google Scholar] [CrossRef]
- Alcindor, T.; Beauger, N. Oxaliplatin: A review in the era of molecularly targeted therapy. Curr. Oncol. 2011, 18, 18–25. [Google Scholar] [CrossRef]
- Passardi, A.; Canale, M.; Valgiusti, M.; Ulivi, P. Immune Checkpoints as a Target for Colorectal Cancer Treatment. Int. J. Mol. Sci. 2017, 18, 1324. [Google Scholar] [CrossRef]
- Bartnik, A.; Nirmal, A.J.; Yang, S.Y. Peptide Vaccine Therapy in Colorectal Cancer. Vaccines 2012, 1, 1–16. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Berners-Price, S.J. Gold-Based Therapeutic Agents: A New Perspective. In Bioinorganic Medicinal Chemistry; Alessio, E., Ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2011; pp. 197–221. [Google Scholar]
- Bindoli, A.; Rigobello, M.P.; Scutari, G.; Gabbiani, C.; Casini, A.; Messori, L. Thioredoxin reductase: A target for gold compounds acting as potential anticancer drugs. Coord. Chem. Rev. 2009, 253, 1692–1707. [Google Scholar] [CrossRef]
- Casini, A. Exploring the mechanisms of metal-based pharmacological agents via an integrated approach. J. Inorg. Biochem. 2012, 109, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, Q.; Xiao, L.; Tang, D.; Dou, Q.P.; Liu, J. Metal-based proteasomal deubiquitinase inhibitors as potential anticancer agents. Cancer Metastasis Rev. 2017, 36, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Nardon, C.; Boscutti, G.; Fregona, D. Beyond Platinums: Gold Complexes as Anticancer Agents. Anticancer Res. 2014, 34, 487–492. [Google Scholar]
- Casini, A.; Messori, L. Molecular mechanisms and proposed targets for selected anticancer gold compounds. Curr. Top. Med. Chem. 2011, 11, 2647–2660. [Google Scholar] [CrossRef] [PubMed]
- Berglof, F.E.; Berglof, K.; Walz, D.T. Auranofin-oral chryso-therapeutic agent for treatment of rheumatoid-arthritis. J. Rheumatol. 1978, 5, 68–74. [Google Scholar]
- Glennas, A.; Kvien, T.K.; Andrup, O.; ClarkeJenssen, O.; Karstensen, B.; Brodin, U. Auranofin is safe and superior to placebo in elderly-onset rheumatoid arthritis. Br. J. Rheumatol. 1997, 36, 870–877. [Google Scholar] [CrossRef]
- Madeira, J.M.; Gibson, D.L.; Kean, W.F.; Klegeris, A. The biological activity of auranofin: Implications for novel treatment of diseases. Inflammopharmacology 2012, 20, 297–306. [Google Scholar] [CrossRef]
- Nobili, S.; Mini, E.; Landini, I.; Gabbiani, C.; Casini, A.; Messori, L. Gold compounds as anticancer agents: Chemistry, cellular pharmacology, and preclinical studies. Med. Res. Rev. 2010, 30, 550–580. [Google Scholar] [CrossRef]
- University of Kansas. Phase I and II Study of Auranofin in Chronic Lymphocytic Leukemia (CLL). [ClinicalTrials.Gov Identifier NCT01419691]; US National Institutes of Health, ClinicalTrials.gov.: Bethesda, MD, USA, 2016.
- Majo Clinic Cancer Center. Auranofin in Treating Patients with Recurrent Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. [ClinicalTrials.Gov Identifier NCT01747798]; US National Institutes of Health, ClinicalTrials.gov.: Bethesda, MD, USA, 2018.
- McKeage, M.J.; Maharaj, L.; Berners-Price, S.J. Mechanisms of cytotoxicity and antitumor activity of gold(I) phosphine complexes: The possible role of mitochondria. Coord. Chem. Rev. 2002, 232, 127–135. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Messori, L.; Marcon, G.; Agostina Cinellu, M.; Bragadin, M.; Folda, A.; Scutari, G.; Bindoli, A. Gold complexes inhibit mitochondrial thioredoxin reductase: Consequences on mitochondrial functions. J. Inorg. Biochem. 2004, 98, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, H.J.; Huang, Q.; Lu, L.; Min, W. Novel action and mechanism of auranofin in inhibition of vascular endothelial growth factor receptor-3-dependent lymphangiogenesis. Anticancer Agents Med. Chem. 2014, 14, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Huang, H.; Dou, Q.P.; Liu, J. Inhibition of 19S proteasome-associated deubiquitinases by metal-containing compounds. Oncoscience 2015, 2, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Fernandes, A.P.; Rigobello, M.P.; Dani, B.; Sorrentino, F.; Tisato, F.; Bjornstedt, M.; Bindoli, A.; Sturaro, A.; Rella, R.; et al. Cancer cell death induced by phosphine gold(I) compounds targeting thioredoxin reductase. Biochem. Pharmacol. 2010, 79, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, H.; You, Y.; Ott, I. Comparative studies on the cytotoxicity, cellular and nuclear uptake of a series of chloro gold(I) phosphine complexes. Polyhedron 2010, 29, 66–69. [Google Scholar] [CrossRef]
- Garcia-Moreno, E.; Gascon, S.; Atrian-Blasco, E.; Rodriguez-Yoldi, M.J.; Cerrada, E.; Laguna, M. Gold(I) complexes with alkylated PTA (1,3,5-triaza-7-phosphaadamantane) phosphanes as anticancer metallodrugs. Eur. J. Med. Chem. 2014, 79, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Atrián-Blasco, E.; Gascón, S.; Rodríguez-Yoldi, M.J.; Laguna, M.; Cerrada, E. Synthesis of Gold(I) Derivatives Bearing Alkylated 1,3,5-Triaza-7-phosphaadamantane as Selective Anticancer Metallodrugs. Eur. J. Inorg. Chem. 2016, 2016, 2791–2803. [Google Scholar] [CrossRef]
- Osterloh, J.; Vicente, M.G.H. Mechanisms of porphyrinoid localization in tumors. J. Porphyr. Phthalocyanines 2002, 6, 305–324. [Google Scholar] [CrossRef]
- Tasan, S.; Licona, C.; Doulain, P.E.; Michelin, C.; Gros, C.P.; Le Gendre, P.; Harvey, P.D.; Paul, C.; Gaiddon, C.; Bodio, E. Gold-phosphine-porphyrin as potential metal-based theranostics. J. Biol. Inorg. Chem. 2015, 20, 143–154. [Google Scholar] [CrossRef]
- Trommenschlager, A.; Chotard, F.; Bertrand, B.; Amor, S.; Dondaine, L.; Picquet, M.; Richard, P.; Bettaieb, A.; Le Gendre, P.; Paul, C.; et al. Gold(I)-BODIPY-imidazole bimetallic complexes as new potential anti-inflammatory and anticancer trackable agents. Dalton Trans. 2017, 46, 8051–8056. [Google Scholar] [CrossRef] [PubMed]
- Santini, C.; Pellei, M.; Papini, G.; Morresi, B.; Galassi, R.; Ricci, S.; Tisato, F.; Porchia, M.; Rigobello, M.P.; Gandin, V.; et al. In vitro antitumour activity of water soluble Cu(I), Ag(I) and Au(I) complexes supported by hydrophilic alkyl phosphine ligands. J. Inorg. Biochem. 2011, 105, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Berners-Price, S.J.; Mirabelli, C.K.; Johnson, R.K.; Mattern, M.R.; McCabe, F.L.; Faucette, L.F.; Sung, C.M.; Mong, S.M.; Sadler, P.J.; Crooke, S.T. In vivo antitumor-activity and invitro cytotoxic properties of bis 1,2-bis(diphenylphosphino)ethane gold(I) chloride. Cancer Res. 1986, 46, 5486–5493. [Google Scholar] [PubMed]
- Caruso, F.; Rossi, M.; Tanski, J.; Pettinari, C.; Marchetti, F. Antitumor activity of the mixed phosphine gold species chlorotriphenylphosphine-1,3-bis(diphenylphosphino)propanegold(I). J. Med. Chem. 2003, 46, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Lupidi, G.; Avenali, L.; Bramucci, M.; Quassinti, L.; Pettinari, R.; Khalife, H.K.; Gali-Muhtasib, H.; Marchetti, F.; Pettinari, C. Synthesis, properties, and antitumor effects of a new mixed phosphine gold(I) compound in human colon cancer cells. J. Inorg. Biochem. 2013, 124, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Hoke, G.D.; Macia, R.A.; Meunier, P.C.; Bugelski, P.J.; Mirabelli, C.K.; Rush, G.F.; Matthews, W.D. Invivo And Invitro Cardiotoxicity Of A Gold-Containing Antineoplastic Drug Candidate In The Rabbit. Toxicol. Appl. Pharmacol. 1989, 100, 293–306. [Google Scholar] [CrossRef]
- Wetzel, C.; Kunz, P.C.; Kassack, M.U.; Hamacher, A.; Böhler, P.; Watjen, W.; Ott, I.; Rubbiani, R.; Spingler, B. Gold(i) complexes of water-soluble diphos-type ligands: Synthesis, anticancer activity, apoptosis and thioredoxin reductase inhibition. Dalton Trans. 2011, 40, 9212–9220. [Google Scholar] [CrossRef]
- Hayashi, R.; Nakatsui, K.; Sugiyama, D.; Kitajima, T.; Oohara, N.; Sugiya, M.; Osada, S.; Kodama, H. Anti-tumor activities of Au(I) complexed with bisphosphines in HL-60 cells. J. Inorg. Biochem. 2014, 137, 109–114. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Cao, R.; Zhang, W.; Yin, M.; Xiao, X.; Liu, Q.; Huang, N. A Soluble Bis-Chelated Gold(I) Diphosphine Compound with Strong Anticancer Activity and Low Toxicity. J. Med. Chem. 2013, 56, 1455–1466. [Google Scholar] [CrossRef]
- Horvath, U.E.I.; Dobrzańska, L.; Strasser, C.E.; Bouwer, W.; Joone, G.; van Rensburg, C.E.J.; Cronje, S.; Raubenheimer, H.G. Amides of gold(I) diphosphines prepared from N-heterocyclic sources and their in vitro and in vivo screening for anticancer activity. J. Inorg. Biochem. 2012, 111, 80–90. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Gold derivatives for the treatment of cancer. Crit. Rev. Oncol. Hemathol. 2002, 42, 225–248. [Google Scholar] [CrossRef]
- Serebryanskaya, T.V.; Lyakhov, A.S.; Ivashkevich, L.S.; Schur, J.; Frias, C.; Prokop, A.; Ott, I. Gold(I) thiotetrazolates as thioredoxin reductase inhibitors and antiproliferative agents. Dalton Trans. 2015, 44, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Ott, I.; Xu, Y.F.; Liu, J.W.; Kokoschka, M.; Harlos, M.; Sheldrick, W.S.; Qian, X.H. Sulfur-substituted naphthalimides as photoactivatable anticancer agents: DNA interaction, fluorescence imaging, and phototoxic effects in cultured tumor cells. Bioorg. Med. Chem. 2008, 16, 7107–7116. [Google Scholar] [CrossRef] [PubMed]
- Bagowski, C.P.; You, Y.; Scheffler, H.; Vlecken, D.H.; Schmitza, D.J.; Ott, I. Naphthalimide gold(I) phosphine complexes as anticancer metallodrugs. Dalton Trans. 2009, 48, 10799–10805. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.I.; Ooi, K.K.; Akim, A.M.; Ang, K.P.; Fairuz, Z.A.; Halim, S.; Ng, S.W.; Seng, H.L.; Tiekink, E.R.T. The influence of R substituents in triphenylphosphinegold(I) carbonimidothioates, Ph3PAu SC(OR) = NPh (R = Me, Et and iPr), upon in vitro cytotoxicity against the HT-29 colon cancer cell line and upon apoptotic pathways. J. Inorg. Biochem. 2013, 127, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Ooi, K.K.; Yeo, C.I.; Ang, K.P.; Akim, A.M.; Cheah, Y.K.; Halim, S.N.; Seng, H.L.; Tiekink, E.R. Phosphanegold(I) thiolates, Ph3PAu[SC(OR)=NC 6H 4Me-4] for R = Me, Et and iPr, induce apoptosis, cell cycle arrest and inhibit cell invasion of HT-29 colon cancer cells through modulation of the nuclear factor-kappaB activation pathway and ubiquitination. J. Biol. Inorg. Chem. 2015, 20, 855–873. [Google Scholar] [CrossRef]
- Altaf, M.; Monim-ul-Mehboob, M.; Isab, A.A.; Dhuna, V.; Bhatia, G.; Dhuna, K.; Altuwaijri, S. The synthesis, spectroscopic characterization and anticancer activity of new mono and binuclear phosphanegold(I) dithiocarbamate complexes. New J. Chem. 2015, 39, 377–385. [Google Scholar] [CrossRef]
- Miranda, S.; Vergara, E.; Mohr, F.; de Vos, D.; Cerrada, E.; Mendia, A.; Laguna, M. Synthesis, characterization, and in vitro cytotoxicity of some gold(I) and trans platinum(II) thionate complexes containing water-soluble PTA and DAPTA ligands. X-ray crystal structures of Au(SC4H3N2)(PTA), trans- Pt(SC4H3N2)(2)(PTA)(2), trans- Pt(SC5H4N)(2)(PTA)(2), and trans- Pt(SC5H4N)(2)(DAPTA)(2). Inorg. Chem. 2008, 47, 5641–5648. [Google Scholar]
- Atrian-Blasco, E.; Gascon, S.; Rodriguez-Yoldi, M.J.; Laguna, M.; Cerrada, E. Novel Gold(I) Thiolate Derivatives Synergistic with 5-Fluorouracil as Potential Selective Anticancer Agents in Colon Cancer. Inorg. Chem. 2017, 56, 8562–8579. [Google Scholar] [CrossRef]
- García-Moreno, E.; Gascón, S.; Jalón, J.A.G.d.; Romanos, E.; Rodriguez-Yoldi, M.J.; Cerrada, E.; Laguna, M. In vivo anticancer activity, toxicology and histopathological studies of the thiolate gold(I) complex [Au(Spyrimidine)(PTA-CH2Ph)]. Br. Anticancer Agents Med. Chem. 2015, 15, 773–782. [Google Scholar] [CrossRef]
- Chaves, J.D.S.; Neumann, F.; Francisco, T.M.; Corrêa, C.C.; Lopes, M.T.P.; Silva, H.; Fontes, A.P.S.; de Almeida, M.V. Synthesis and cytotoxic activity of gold(I) complexes containing phosphines and 3-benzyl-1,3-thiazolidine-2-thione or 5-phenyl-1,3,4-oxadiazole-2-thione as ligands. Inorg. Chim. Acta 2014, 414, 85–90. [Google Scholar] [CrossRef]
- de Almeida, A.M.; de Oliveira, B.A.; de Castro, P.P.; de Mendonca, C.C.; Furtado, R.A.; Nicolella, H.D.; da Silva, V.L.; Diniz, C.G.; Tavares, D.C.; Silva, H.; et al. Lipophilic gold(I) complexes with 1,3,4-oxadiazol-2-thione or 1,3-thiazolidine-2-thione moieties: Synthesis and their cytotoxic and antimicrobial activities. Biometals 2017, 30, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Machado, R.C.; Grazul, R.M.; Lopes, M.T.P.; Correa, C.C.; Dos Santos, H.F.; de Almeida, M.V.; Silva, H. Novel antitumor adamantane-azole gold(I) complexes as potential inhibitors of thioredoxin reductase. J. Biol. Inorg. Chem. 2016, 21, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Tavares, T.T.; Azevedo, G.C.; Garcia, A.; Carpanez, A.G.; Lewer, P.M.; Paschoal, D.; Muller, B.L.; Dos Santos, H.F.; Matos, R.C.; Silva, H.; et al. Gold(I) complexes with aryl-thiosemicarbazones: Molecular modeling, synthesis, cytotoxicity and TrxR inhibition. Polyhedron 2017, 132, 95–104. [Google Scholar] [CrossRef]
- Navarro, M.; Castro, W.; Gonzalez, S.; Abad, M.J.; Taylor, P. Synthesis and Anticancer Activity of Gold(I)-Chloroquine Complexes. J. Mex. Chem. Soc. 2013, 57, 220–229. [Google Scholar] [CrossRef]
- Liu, W.K.; Gust, R. Update on metal N-heterocyclic carbene complexes as potential anti-tumor Metallodrugs. Coord. Chem. Rev. 2016, 329, 191–213. [Google Scholar] [CrossRef]
- Liu, W.K.; Gust, R. Metal N-heterocyclic carbene complexes as potential antitumor metallodrugs. Chem. Soc. Rev. 2013, 42, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Mora, M.; Gimeno, M.C.; Visbal, R. Recent advances in gold-NHC complexes with biological properties. Chem. Soc. Rev. 2019, 48, 447–462. [Google Scholar] [CrossRef]
- Liu, W.; Bensdorf, K.; Proetto, M.; Abram, U.; Hagenbach, A.; Gust, R. NHC Gold Halide Complexes Derived from 4,5-Diarylimidazoles: Synthesis, Structural Analysis, and Pharmacological Investigations as Potential Antitumor Agents. J. Med. Chem. 2011, 54, 8605–8615. [Google Scholar] [CrossRef]
- Liu, W.K.; Chen, X.H.; Gust, R. Synthesis, Antitumor, and Antibacterial Activity of Bis 4,5-diarylimidazol-2-ylidene methane Derivatives. Arch. Pharm. Chem. Life Sci. 2012, 345, 557–564. [Google Scholar] [CrossRef]
- Kaps, L.; Biersack, B.; Müller-Bunz, H.; Mahal, K.; Münzner, J.; Tacke, M.; Mueller, T.; Schobert, R. Gold(I)–NHC complexes of antitumoral diarylimidazoles: Structures, cellular uptake routes and anticancer activities. J. Inorg. Biochem. 2012, 106, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Muenzner, J.K.; Biersack, B.; Kalie, H.; Andronache, I.C.; Kaps, L.; Schuppan, D.; Sasse, F.; Schobert, R. Gold(I) Biscarbene Complexes Derived from Vascular-Disrupting Combretastatin A-4 Address Different Targets and Show Antimetastatic Potential. Chem. Med. Chem. 2014, 9, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.; Dada, O.; Muller-Bunz, H.; Rothemund, M.; Sanchez-Sanz, G.; Schobert, R.; Zhu, X.M.; Tacke, M. Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A. Molecules 2018, 23, 2031. [Google Scholar] [CrossRef] [PubMed]
- Arcau, J.; Andermark, V.; Rodrigues, M.; Giannicchi, I.; Perez-Garcia, L.; Ott, I.; Rodriguez, L. Synthesis and Biological Activity of Gold(I) N-Heterocyclic Carbene Complexes with Long Aliphatic Side Chains. Eur. J. Inorg. Chem. 2014, 35, 6117–6125. [Google Scholar] [CrossRef]
- Altaf, M.; Monim-ul-Mehboob, M.; Seliman, A.A.A.; Isab, A.A.; Dhuna, V.; Bhatia, G.; Dhuna, K. Synthesis, X-ray structures, spectroscopic analysis and anticancer activity of novel gold(I) carbene complexes. J. Organomet. Chem. 2014, 765, 68–79. [Google Scholar] [CrossRef]
- Seliman, A.A.A.; Altaf, M.; Onawole, A.T.; Ahmad, S.; Ahmed, M.Y.; Al-Saadi, A.A.; Altuwaijri, S.; Bhatia, G.; Singh, J.; Isab, A.A. Synthesis, X-ray structures and anticancer activity of gold(I)-carbene complexes with selenones as co-ligands and their molecular docking studies with thioredoxin reductase. J. Organomet. Chem. 2017, 848, 175–183. [Google Scholar] [CrossRef]
- Seliman, A.A.A.; Altaf, M.; Odewunmi, N.A.; Kawde, A.N.; Zierkiewicz, W.; Ahmad, S.; Altuwaijri, S.; Isab, A.A. Synthesis, X-ray structure, DFT calculations and anticancer activity of a selenourea coordinated gold(I)-carbene complex. Polyhedron 2017, 137, 197–206. [Google Scholar] [CrossRef]
- Rubbiani, R.; Schuh, E.; Meyer, A.; Lemke, J.; Wimberg, J.; Metzler-Nolte, N.; Meyer, F.; Mohr, F.; Ott, I. TrxR inhibition and antiproliferative activities of structurally diverse gold N-heterocyclic carbene complexes. Med. Chem. Commun. 2013, 4, 942–948. [Google Scholar] [CrossRef]
- Pratesi, A.; Cirri, D.; Durovic, M.D.; Pillozzi, S.; Petroni, G.; Bugarcic, Z.D.; Messori, L. New gold carbene complexes as candidate anticancer agents. Biometals 2016, 29, 905–911. [Google Scholar] [CrossRef]
- Fernandez-Gallardo, J.; Elie, B.T.; Sanau, M.; Contel, M. Versatile synthesis of cationic N-heterocyclic carbene-gold(I) complexes containing a second ancillary ligand. Design of heterobimetallic ruthenium-gold anticancer agents. Chem. Commun. 2016, 52, 3155–3158. [Google Scholar] [CrossRef]
- Rubbiani, R.; Kitanovic, I.; Alborzinia, H.; Can, S.; Kitanovic, A.; Onambele, L.A.; Stefanopoulou, M.; Geldmacher, Y.; Sheldrick, W.S.; Wolber, G.; et al. Benzimidazol-2-ylidene Gold(I) Complexes Are Thioredoxin Reductase Inhibitors with Multiple Antitumor Properties. J. Med. Chem. 2010, 53, 8608–8618. [Google Scholar] [CrossRef]
- Cheng, X.L.; Holenya, P.; Can, S.; Alborzinia, H.; Rubbiani, R.; Ott, I.; Wolfl, S. A TrxR inhibiting gold(I) NHC complex induces apoptosis through ASK1-p38-MAPK signaling in pancreatic cancer cells. Mol. Cancer 2014, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Salassa, L.; de Almeida, A.; Casini, A.; Ott, I. Cytotoxic Gold(I) N-heterocyclic Carbene Complexes with Phosphane Ligands as Potent Enzyme Inhibitors. Chem. Med. Chem. 2014, 9, 1205–1210. [Google Scholar] [CrossRef]
- Rubbiani, R.; Can, S.; Kitanovic, I.; Alborzinia, H.; Stefanopoulou, M.; Kokoschka, M.; Monchgesang, S.; Sheldrick, W.S.; Wolfl, S.; Ott, I. Comparative in Vitro Evaluation of N-Heterocyclic Carbene Gold(I) Complexes of the Benzimidazolylidene Type. J. Med. Chem. 2011, 54, 8646–8657. [Google Scholar] [CrossRef]
- Holenya, P.; Can, S.Z.; Rubbiani, R.; Alborzinia, H.; Junger, A.; Cheng, X.L.; Ott, I.; Wolfl, S. Detailed analysis of pro-apoptotic signaling and metabolic adaptation triggered by a N-heterocyclic carbene-gold(I) complex. Metallomics 2014, 6, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Jhulki, L.; Dutta, P.; Santra, M.K.; Cardoso, M.H.; Oshiro, K.G.N.; Franco, O.L.; Bertolasi, V.; Isab, A.A.; Bielawski, C.W.; Dinda, J. Synthesis and cytotoxic characteristics displayed by a series of Ag(i)-, Au(i)- and Au(iii)-complexes supported by a common N-heterocyclic carbene. New J. Chem. 2018, 42, 13948–13956. [Google Scholar] [CrossRef]
- Dinda, J.; Samanta, T.; Nandy, A.; Das Saha, K.; Seth, S.K.; Chattopadhyay, S.K.; Bielawskie, C.W. N-heterocyclic carbene supported Au(I) and Au(III) complexes: A comparison of cytotoxicities. New J. Chem. 2014, 38, 1218–1224. [Google Scholar] [CrossRef]
- Samanta, T.; Munda, R.N.; Roymahapatra, G.; Nandy, A.; Das Saha, K.; Al-Deyab, S.S.; Dinda, J. Silver(I), Gold(I) and Gold(III)-N-Heterocyclic carbene complexes of naphthyl substituted annelated ligand: Synthesis, structure and cytotoxicity. J. Organomet. Chem. 2015, 791, 183–191. [Google Scholar] [CrossRef]
- Meyer, A.; Oehninger, L.; Geldmacher, Y.; Alborzinia, H.; Wolfl, S.; Sheldrick, W.S.; Ott, I. Gold(I) N-Heterocyclic Carbene Complexes with Naphthalimide Ligands as Combined Thioredoxin Reductase Inhibitors and DNA Intercalators. Chem. Med. Chem. 2014, 9, 1794–1800. [Google Scholar] [CrossRef]
- Baron, M.; Bellemin-Laponnaz, S.; Tubaro, C.; Basato, M.; Bogialli, S.; Dolmella, A. Synthesis and biological assays on cancer cells of dinuclear gold complexes with novel functionalised di(N-heterocyclic carbene) ligands. J. Inorg. Biochem. 2014, 141, 94–102. [Google Scholar] [CrossRef]
- Cerrada, E.; Fernández-Moreira, V.; Gimeno, M.C. Gold and platinum alkynyl complexes for biomedical applications. In Advances in Organometallic Chemistry; Pérez, P., Ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2019; Volume 71, pp. 227–258. [Google Scholar]
- Schuh, E.; Valiahdi, S.M.; Jakupec, M.A.; Keppler, B.K.; Chiba, P.; Mohr, F. Synthesis and biological studies of some gold(I) complexes containing functionalised alkynes. Dalton Trans. 2009, 48, 10841–10845. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Bagowski, C.P.; Kokoschka, M.; Stefanopoulou, M.; Alborzinia, H.; Can, S.; Vlecken, D.H.; Sheldrick, W.S.; Wolfl, S.; Ott, I. On the Biological Properties of Alkynyl Phosphine Gold(I) Complexes. Angew. Chem. Int. Ed. 2012, 51, 8895–8899. [Google Scholar] [CrossRef] [PubMed]
- Andermark, V.; Goke, K.; Kokoschka, M.; Abu El Maaty, M.A.; Lum, C.T.; Zou, T.; Sun, R.W.-Y.; Aguilo, E.; Oehninger, L.; Rodriguez, L.; et al. Alkynyl gold(I) phosphane complexes: Evaluation of structure-activity-relationships for the phosphane ligands, effects on key signaling proteins and preliminary in-vivo studies with a nanoformulated complex. J. Inorg. Biochem. 2016, 160, 140–148. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, Z.; Yuan, J.; Zhao, Y.-X.; Qiao, Z.-Y.; Gao, Y.-J.; Yu, G.-A.; Li, J.; Wang, H. Synthesis and Molecular Recognition Studies on Small-Molecule Inhibitors for Thioredoxin Reductase. J. Med. Chem. 2014, 57, 8132–8139. [Google Scholar] [CrossRef] [PubMed]
- Cristina Sánchez-de-Diego, I.M.; Pérez, R.; Gascón, S.; Rodriguez-Yoldi, M.J.; Cerrada, E. The anticancer effect related to disturbances in redox balance on Caco-2 cells caused by an alkynyl gold(I) complex. J. Inorg. Biochem. 2017, 166, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Marmol, I.; Virumbrales-Munoz, M.; Quero, J.; Sanchez-De-Diego, C.; Fernandez, L.; Ochoa, I.; Cerrada, E.; Yoldi, M.J.R. Alkynyl gold(I) complex triggers necroptosis via ROS generation in colorectal carcinoma cells. J. Inorg. Biochem. 2017, 176, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Arcau, J.; Andermark, V.; Aguilo, E.; Gandioso, A.; Moro, A.; Cetina, M.; Lima, J.C.; Rissanen, K.; Ott, I.; Rodriguez, L. Luminescent alkynyl-gold(I) coumarin derivatives and their biological activity. Dalton Trans. 2014, 43, 4426–4436. [Google Scholar] [CrossRef]
- Gavara, R.; Aguilo, E.; Schur, J.; Llorca, J.; Ott, I.; Rodriguez, L. Study of the effect of the chromophore and nuclearity on the aggregation and potential biological activity of gold(I) alkynyl complexes. Inorg. Chim. Acta 2016, 446, 189–197. [Google Scholar] [CrossRef]
- De Nisi, A.; Bergamini, C.; Leonzio, M.; Sartor, G.; Fato, R.; Naldi, M.; Monari, M.; Calonghi, N.; Bandini, M. Synthesis, cytotoxicity and anti-cancer activity of new alkynyl-gold(I) complexes. Dalton Trans. 2016, 45, 1546–1553. [Google Scholar] [CrossRef]
- Garcia-Moreno, E.; Gascon, S.; Rodriguez-Yoldi, M.J.; Cerrada, E.; Laguna, M. S-Propargylthiopyridine Phosphane Derivatives As Anticancer Agents: Characterization and Antitumor Activity. Organometallics 2013, 32, 3710–3720. [Google Scholar] [CrossRef]
- Garcia-Moreno, E.; Tomas, A.; Atrian-Blasco, E.; Gascon, S.; Romanos, E.; Rodriguez-Yoldi, M.J.; Cerrada, E.; Laguna, M. In vitro and in vivo evaluation of organometallic gold(I) derivatives as anticancer agents. Dalton Trans. 2016, 45, 2462–2475. [Google Scholar] [CrossRef] [PubMed]
- Vanicek, S.; Kopacka, H.; Wurst, K.; Vergeiner, S.; Kankowski, S.; Schur, J.; Bildstein, B.; Ott, I. Cobaltoceniumethynyl gold(I) as an unusual heterodinuclear bioorganometallic fragment to study the biological properties of alkynyl gold complexes. Dalton Trans. 2016, 45, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Gutiérrez, A.; Ott, I.; Rodríguez, L. Phosphine-bridged dinuclear gold(I) alkynyl complexes: Thioredoxin reductase inhibition and cytotoxicity. Inorg. Chim. Acta 2013, 398, 72–76. [Google Scholar] [CrossRef]
- Langdon-Jones, E.E.; Lloyd, D.; Hayes, A.J.; Wainwright, S.D.; Mottram, H.J.; Coles, S.J.; Horton, P.N.; Pope, S.J.A. Alkynyl-naphthalimide Fluorophores: Gold Coordination Chemistry and Cellular Imaging Applications. Inorg. Chem. 2015, 54, 6606–6615. [Google Scholar] [CrossRef] [PubMed]
- Hossen, S.; Hossain, M.K.; Basher, M.K.; Mia, M.N.H.; Rahman, M.T.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, F.; Xiong, F.; Gu, N. The Smart Drug Delivery System and Its Clinical Potential. Theranostics 2016, 6, 1306–1323. [Google Scholar] [CrossRef] [PubMed]
- Kalaydina, R.V.; Bajwa, K.; Qorri, B.; Decarlo, A.; Szewczuk, M.R. Recent advances in “smart” delivery systems for extended drug release in cancer therapy. Int. J. Nanomed. 2018, 13, 4727–4745. [Google Scholar] [CrossRef] [PubMed]
- Upreti, M.; Jyoti, A.; Sethi, P. Tumor microenvironment and nanotherapeutics. Transl. Cancer Res. 2013, 2, 309–319. [Google Scholar]
- Singh, P.; Pandit, S.; Mokkapati, V.; Garg, A.; Ravikumar, V.; Mijakovic, I. Gold Nanoparticles in Diagnostics and Therapeutics for Human Cancer. Int. J. Mol. Sci. 2018, 19, 1979. [Google Scholar] [CrossRef]
- Wozniak, A.; Malankowska, A.; Nowaczyk, G.; Grzeskowiak, B.F.; Tusnio, K.; Slomski, R.; Zaleska-Medynska, A.; Jurga, S. Size and shape-dependent cytotoxicity profile of gold nanoparticles for biomedical applications. J. Mater. Sci. Mater. Med. 2017, 28, 92. [Google Scholar] [CrossRef]
- Steckiewicz, K.P.; Barcinska, E.; Malankowska, A.; Zauszkiewicz-Pawlak, A.; Nowaczyk, G.; Zaleska-Medynska, A.; Inkielewicz-Stepniak, I. Impact of gold nanoparticles shape on their cytotoxicity against human osteoblast and osteosarcoma in in vitro model. Evaluation of the safety of use and anti-cancer potential. J. Mater. Sci. Mater. Med. 2019, 30, 22. [Google Scholar] [CrossRef] [PubMed]
- Patra, C.R.; Bhattacharya, R.; Mukhopadhyay, D.; Mukherjee, P. Fabrication of gold nanoparticles for targeted therapy in pancreatic cancer. Adv. Drug Deliv Rev. 2010, 62, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Dreaden, E.C.; Austin, L.A.; Mackey, M.A.; El-Sayed, M.A. Size matters: Gold nanoparticles in targeted cancer drug delivery. Ther. Deliv. 2012, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Mercado-Lubo, R.; Zhang, Y.; Zhao, L.; Rossi, K.; Wu, X.; Zou, Y.; Castillo, A.; Leonard, J.; Bortell, R.; Greiner, D.L.; et al. A Salmonella nanoparticle mimic overcomes multidrug resistance in tumours. Nat. Commun. 2016, 7, 12225. [Google Scholar] [CrossRef] [PubMed]
- Arya, M.; Singh, P.; Tripathi, C.B.; Parashar, P.; Singh, M.; Kanoujia, J.; Guleria, A.; Kaithwas, G.; Gupta, K.P.; Saraf, S.A. Pectin-encrusted gold nanocomposites containing phytic acid and jacalin: 1,2-dimethylhydrazine-induced colon carcinogenesis in Wistar rats, PI3K/Akt, COX-2, and serum metabolomics as potential targets. Drug Deliv. Transl. Res. 2019, 9, 53–65. [Google Scholar] [CrossRef]
- Li, J.; Yoong, S.L.; Goh, W.J.; Czarny, B.; Yang, Z.; Poddar, K.; Dykas, M.M.; Patra, A.; Venkatesan, T.; Panczyk, T.; et al. In vitro controlled release of cisplatin from gold-carbon nanobottles via cleavable linkages. Int. J. Nanomed. 2015, 10, 7425–7441. [Google Scholar]
- Fan, L.; Campagnoli, S.; Wu, H.; Grandi, A.; Parri, M.; De Camilli, E.; Grandi, G.; Viale, G.; Pileri, P.; Grifantini, R.; et al. Negatively charged AuNP modified with monoclonal antibody against novel tumor antigen FAT1 for tumor targeting. J. Exp. Clin. Cancer Res. 2015, 34, 103. [Google Scholar] [CrossRef]
- Mayer, K.M.; Hafner, J.H. Localized surface plasmon resonance sensors. Chem. Rev. 2011, 111, 3828–3857. [Google Scholar] [CrossRef]
- Maier, S.A. Localized surface plasmons. In Plasmonics: Fundamentals and Application, 1st ed.; Springer: New York, NY, USA, 2007; pp. 65–88. [Google Scholar]
- Amendola, V.; Pilot, R.; Frasconi, M.; Marago, O.M.; Iati, M.A. Surface plasmon resonance in gold nanoparticles: A review. J. Phys. Condens. Matter 2017, 29, 203002. [Google Scholar] [CrossRef]
- Huang, X.; El-Sayed, M.A. Gold nanoparticles: Optical properties and implementations in cancer diagnosis and photothermal therapy. J. Adv. Res. 2010, 1, 13–28. [Google Scholar] [CrossRef]
- Mie, G. Contributions to the Optics of Turbid Media, Particularly of Colloidal Metal Solutions. Ann. Phys. 1908, 25, 377–445. [Google Scholar] [CrossRef]
- Haes, A.J.; Haynes, C.L.; McFarland, A.D.; Schatz, G.C.; Van Duyne, R.P.; Zou, S. Plasmonic Materials for Surface-Enhanced Sensing and Spectroscopy. MRS Bull. 2005, 30, 368–375. [Google Scholar] [CrossRef]
- Kreibig, U.; Vollmer, M. Experimental results and discussion. In Optical Properties of Metal Clusters, 1st ed.; Springer-Verlag: Berlin, Germany, 1995; pp. 275–436. [Google Scholar]
- Jain, P.K.; Lee, K.S.; El-Sayed, I.H.; El-Sayed, M.A. Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: Applications in biological imaging and biomedicine. J. Phys. Chem. B 2006, 110, 7238–7248. [Google Scholar] [CrossRef] [PubMed]
- Link, S.; El-Sayed, M.A. Optical properties and ultrafast dynamics of metallic nanocrystals. Annu. Rev. Phys. Chem. 2003, 54, 331–366. [Google Scholar] [CrossRef] [PubMed]
- Alabastri, A.; Tuccio, S.; Giugni, A.; Toma, A.; Liberale, C.; Das, G.; Angelis, F.; Fabrizio, E.D.; Zaccaria, R.P. Molding of Plasmonic Resonances in Metallic Nanostructures: Dependence of the Non-Linear Electric Permittivity on System Size and Temperature. Materials 2013, 6, 4879–4910. [Google Scholar] [CrossRef]
- Amendola, V.; Saija, R.; Marago, O.M.; Iati, M.A. Superior plasmon absorption in iron-doped gold nanoparticles. Nanoscale 2015, 7, 8782–8792. [Google Scholar] [CrossRef]
- Poletti, A.; Fracasso, G.; Conti, G.; Pilot, R.; Amendola, V. Laser generated gold nanocorals with broadband plasmon absorption for photothermal applications. Nanoscale 2015, 7, 13702–13714. [Google Scholar] [CrossRef] [PubMed]
- Hainfeld, J.F.; Slatkin, D.N.; Focella, T.M.; Smilowitz, H.M. Gold nanoparticles: A new X-ray contrast agent. Br. J. Radiol. 2006, 79, 248–253. [Google Scholar] [CrossRef]
- Nguyen, V.P.; Li, Y.; Qian, W.; Liu, B.; Tian, C.; Zhang, W.; Huang, Z.; Ponduri, A.; Tarnowski, M.; Wang, X.; et al. Contrast Agent Enhanced Multimodal Photoacoustic Microscopy and Optical Coherence Tomography for Imaging of Rabbit Choroidal and Retinal Vessels in vivo. Sci. Rep. 2019, 9, 5945. [Google Scholar] [CrossRef]
- Alric, C.; Taleb, J.; Le Duc, G.; Mandon, C.; Billotey, C.; Le Meur-Herland, A.; Brochard, T.; Vocanson, F.; Janier, M.; Perriat, P.; et al. Gadolinium chelate coated gold nanoparticles as contrast agents for both X-ray computed tomography and magnetic resonance imaging. J. Am. Chem. Soc. 2008, 130, 5908–5915. [Google Scholar] [CrossRef]
- Liu, J.; Xiong, Z.; Zhang, J.; Peng, C.; Klajnert-Maculewicz, B.; Shen, M.; Shi, X. Zwitterionic Gadolinium(III)-Complexed Dendrimer-Entrapped Gold Nanoparticles for Enhanced Computed Tomography/Magnetic Resonance Imaging of Lung Cancer Metastasis. ACS Appl. Mater. Interfaces 2019, 11, 15212–15221. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Shao, Y.; He, H.; Liu, H.; Shen, Y.; Huang, W.; Li, L. Nanoamplifiers synthesized from gadolinium and gold nanocomposites for magnetic resonance imaging. Nanoscale 2013, 5, 3322–3329. [Google Scholar] [CrossRef] [PubMed]
- Doughty AC, V.; Hoover, A.R.; Layton, E.; Murray, C.K.; Howard, E.W.; Chen, W.R. Nanomaterial Applications in Photothermal Therapy for Cancer. Materials 2019, 12, 779. [Google Scholar] [CrossRef] [PubMed]
- Ban, Q.; Bai, T.; Duan, X.; Kong, J. Noninvasive photothermal cancer therapy nanoplatforms via integrating nanomaterials and functional polymers. Biomater. Sci. 2017, 5, 190–210. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.S.; Day, E.S. Gold nanoparticle-mediated photothermal therapy: Applications and opportunities for multimodal cancer treatment. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1449. [Google Scholar] [CrossRef] [PubMed]
- Grosges, T.; Barchiesi, D. Gold Nanoparticles as a Photothermal Agent in Cancer Therapy: The Thermal Ablation Characteristic Length. Molecules 2018, 23, 1316. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Crawford, B.M.; Vo-Dinh, T. Gold nanoparticles-mediated photothermal therapy and immunotherapy. Immunotherapy 2018, 10, 1175–1188. [Google Scholar] [CrossRef] [PubMed]
- Plan Sangnier, A.; Aufaure, R.; Motte, L.; Wilhelm, C.; Guenin, E.; Lalatonne, Y. Hybrid Au@alendronate nanoparticles as dual chemo-photothermal agent for combined cancer treatment. Beilstein J. Nanotechnol. 2018, 9, 2947–2952. [Google Scholar] [CrossRef]
- Emami, F.; Banstola, A.; Vatanara, A.; Lee, S.; Kim, J.O.; Jeong, J.H.; Yook, S. Doxorubicin and Anti-PD-L1 Antibody Conjugated Gold Nanoparticles for Colorectal Cancer Photochemotherapy. Mol. Pharm. 2019, 16, 1184–1199. [Google Scholar] [CrossRef]
- Zou, L.; Wang, H.; He, B.; Zeng, L.; Tan, T.; Cao, H.; He, X.; Zhang, Z.; Guo, S.; Li, Y. Current Approaches of Photothermal Therapy in Treating Cancer Metastasis with Nanotherapeutics. Theranostics 2016, 6, 762–772. [Google Scholar] [CrossRef]
- Nam, J.; Son, S.; Ochyl, L.J.; Kuai, R.; Schwendeman, A.; Moon, J.J. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat. Commun. 2018, 9, 1074. [Google Scholar] [CrossRef] [PubMed]
- Mulens-Arias, V.; Nicolas-Boluda, A.; Gehanno, A.; Balfourier, A.; Carn, F.; Gazeau, F. Polyethyleneimine-assisted one-pot synthesis of quasi-fractal plasmonic gold nanocomposites as a photothermal theranostic agent. Nanoscale 2019, 11, 3344–3359. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Huh, M.; Lee, S.J.; Koo, H.; Kwon, I.C.; Jeong, S.Y.; Kim, K. Photosensitizer-conjugated human serum albumin nanoparticles for effective photodynamic therapy. Theranostics 2011, 1, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Potara, M.; Gabudean, A.M.; Licarete, E.; Banciu, M.; Astilean, S. Designing Theranostic Agents Based on Pluronic Stabilized Gold Nanoaggregates Loaded with Methylene Blue for Multimodal Cell Imaging and Enhanced Photodynamic Therapy. ACS Appl. Mater. Interfaces 2015, 7, 16191–16201. [Google Scholar] [CrossRef] [PubMed]
- Obaid, G.; Chambrier, I.; Cook, M.J.; Russell, D.A. Cancer targeting with biomolecules: A comparative study of photodynamic therapy efficacy using antibody or lectin conjugated phthalocyanine-PEG gold nanoparticles. Photochem. Photobiol. Sci. 2015, 14, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.O.; Ha, K.S. New insights into the mechanisms for photodynamic therapy-induced cancer cell death. Int. Rev. Cell Mol. Biol. 2012, 295, 139–174. [Google Scholar]
- Allison, R.; Moghissi, K.; Downie, G.; Dixon, K. Photodynamic therapy (PDT) for lung cancer. Photodiagn. Photodyn. Ther. 2011, 8, 231–239. [Google Scholar] [CrossRef]
- Kostovic, K.; Pastar, Z.; Ceovic, R.; Mokos, Z.B.; Buzina, D.S.; Stanimirovic, A. Photodynamic therapy in dermatology: Current treatments and implications. Coll. Antropol. 2012, 36, 1477–1481. [Google Scholar]
- Gondivkar, S.M.; Gadbail, A.R.; Choudhary, M.G.; Vedpathak, P.R.; Likhitkar, M.S. Photodynamic treatment outcomes of potentially-malignant lesions and malignancies of the head and neck region: A systematic review. J. Investig. Clin. Dent. 2018, 9, e12270. [Google Scholar] [CrossRef]
- McKenzie, E.; Zaki, P.; Raman, S.; Olson, R.; McFarlane, T.; DeAngelis, C.; Chan, S.; Pidduck, W.; Razvi, Y.; Bushehri, A.; et al. Radiation-induced nausea and vomiting: A comparison between MASCC/ESMO, ASCO, and NCCN antiemetic guidelines. Support. Care Cancer 2019, 27, 783–791. [Google Scholar] [CrossRef]
- Yee, C.; Wang, K.; Asthana, R.; Drost, L.; Lam, H.; Lee, J.; Vesprini, D.; Leung, E.; DeAngelis, C.; Chow, E. Radiation-induced Skin Toxicity in Breast Cancer Patients: A Systematic Review of Randomized Trials. Clin. Breast Cancer 2018, 18, e825–e840. [Google Scholar] [CrossRef] [PubMed]
- Maria, O.M.; Eliopoulos, N.; Muanza, T. Radiation-Induced Oral Mucositis. Front. Oncol 2017, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Hirst, D.G.; O’Sullivan, J.M. Gold nanoparticles as novel agents for cancer therapy. Br. J. Radiol. 2012, 85, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.; Connolly, C.; Schettino, G.; Butterworth, K.T.; Prise, K.M. Biological mechanisms of gold nanoparticle radiosensitization. Cancer Nanotechnol. 2017, 8, 2. [Google Scholar] [CrossRef]
- Penninckx, S.; Heuskin, A.C.; Michiels, C.; Lucas, S. The role of thioredoxin reductase in gold nanoparticle radiosensitization effects. Nanomedicine 2018, 13, 2917–2937. [Google Scholar] [CrossRef] [PubMed]
- Penninckx, S.; Heuskin, A.C.; Michiels, C.; Lucas, S. Thioredoxin Reductase Activity Predicts Gold Nanoparticle Radiosensitization Effect. Nanomaterials 2019, 9, 295. [Google Scholar] [CrossRef]
- Hau, H.; Khanal, D.; Rogers, L.; Suchowerska, N.; Kumar, R.; Sridhar, S.; McKenzie, D.; Chrzanowski, W. Dose enhancement and cytotoxicity of gold nanoparticles in colon cancer cells when irradiated with kilo- and mega-voltage radiation. Bioeng. Transl. Med. 2016, 1, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Saberi, A.; Shahbazi-Gahrouei, D.; Abbasian, M.; Fesharaki, M.; Baharlouei, A.; Arab-Bafrani, Z. Gold nanoparticles in combination with megavoltage radiation energy increased radiosensitization and apoptosis in colon cancer HT-29 cells. Int. J. Radiat. Biol. 2017, 93, 315–323. [Google Scholar] [CrossRef]
- Rezaee, Z.; Yadollahpour, A.; Bayati, V.; Negad Dehbashi, F. Gold nanoparticles and electroporation impose both separate and synergistic radiosensitizing effects in HT-29 tumor cells: An in vitro study. Int. J. Nanomed. 2017, 12, 1431–1439. [Google Scholar] [CrossRef]
- Mirrahimi, M.; Khateri, M.; Beik, J.; Ghoreishi, F.S.; Dezfuli, A.S.; Ghaznavi, H.; Shakeri-Zadeh, A. Enhancement of chemoradiation by co-incorporation of gold nanoparticles and cisplatin into alginate hydrogel. J. Biomed. Mater. Res. B Appl. Biomater. 2019. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, E.J.; Kim, J.W.; Chung, U.S.; Koh, W.G.; Keum, K.C.; Koom, W.S. Gold nanoparticles enhance anti-tumor effect of radiotherapy to hypoxic tumor. Radiat. Oncol. J. 2016, 34, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R.; Overgaard, J. The impact of hypoxia and its modification of the outcome of radiotherapy. J. Radiat. Res. 2016, 57, i90–i98. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mármol, I.; Quero, J.; Rodríguez-Yoldi, M.J.; Cerrada, E. Gold as a Possible Alternative to Platinum-Based Chemotherapy for Colon Cancer Treatment. Cancers 2019, 11, 780. https://doi.org/10.3390/cancers11060780
Mármol I, Quero J, Rodríguez-Yoldi MJ, Cerrada E. Gold as a Possible Alternative to Platinum-Based Chemotherapy for Colon Cancer Treatment. Cancers. 2019; 11(6):780. https://doi.org/10.3390/cancers11060780
Chicago/Turabian StyleMármol, Inés, Javier Quero, María Jesús Rodríguez-Yoldi, and Elena Cerrada. 2019. "Gold as a Possible Alternative to Platinum-Based Chemotherapy for Colon Cancer Treatment" Cancers 11, no. 6: 780. https://doi.org/10.3390/cancers11060780
APA StyleMármol, I., Quero, J., Rodríguez-Yoldi, M. J., & Cerrada, E. (2019). Gold as a Possible Alternative to Platinum-Based Chemotherapy for Colon Cancer Treatment. Cancers, 11(6), 780. https://doi.org/10.3390/cancers11060780