Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment


{kind=link}
{kind=link}
Abstract
1. Tumor Microenvironment and Metastasis
2. ECM Stiffness and Cancer
3. LOX Family Members and Their Role in Development
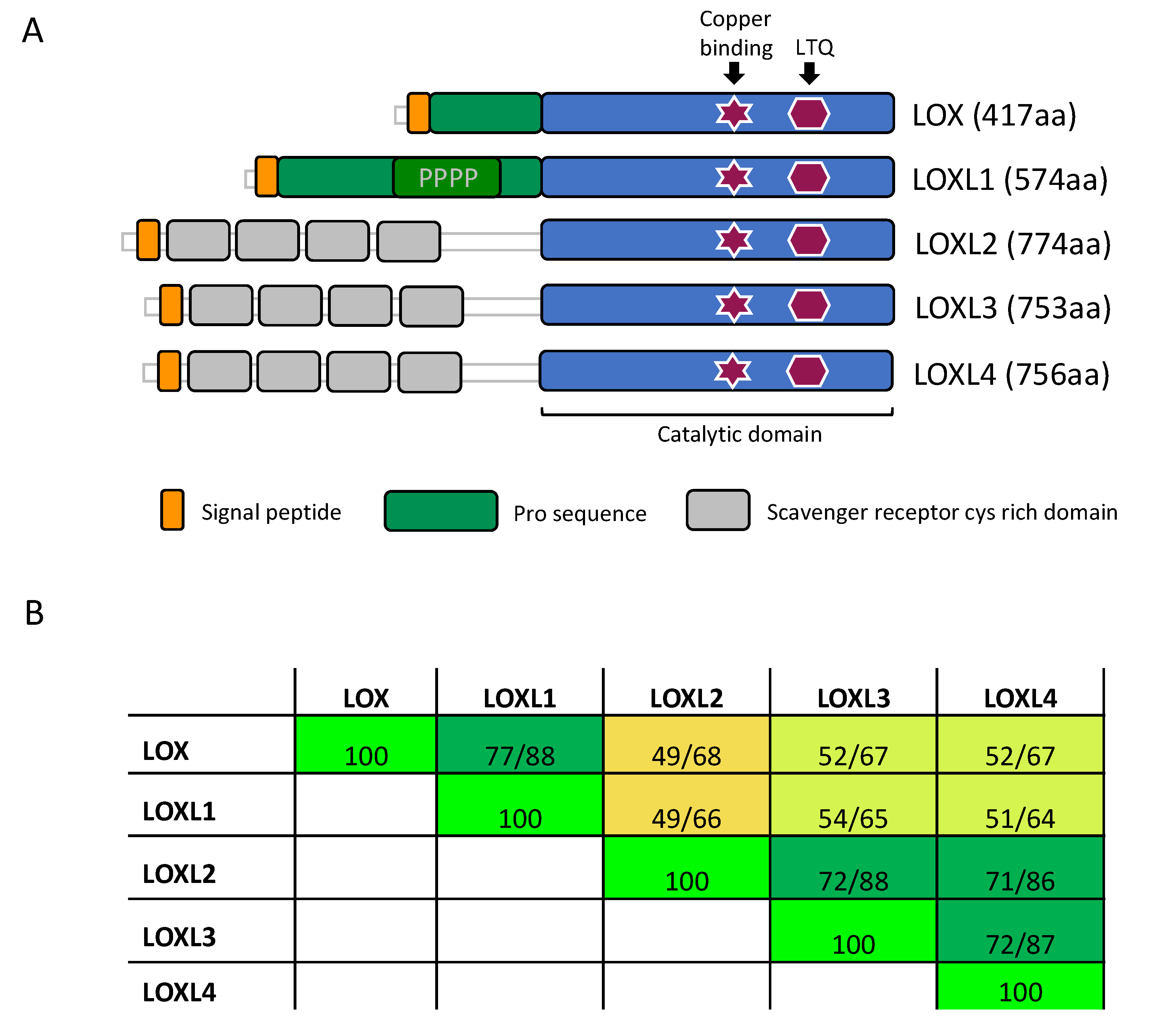
3.1. Structure of LOX Enzymes
3.2. LOX Enzymes in Cancer
4. Matrix Stiffness and Integrin Signaling
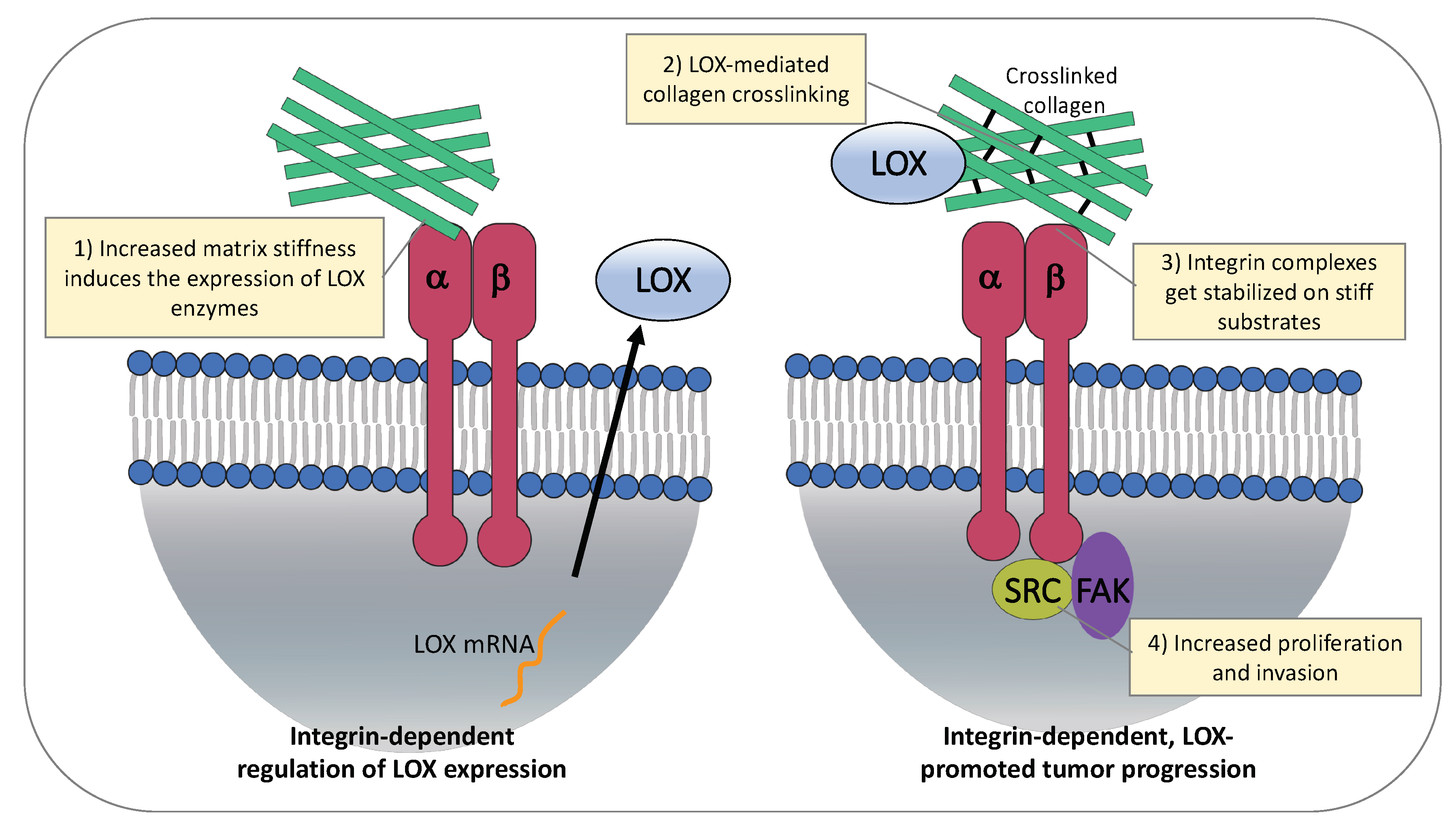
4.1. ECM Stiffness Regulates Expression of LOX Enzymes
4.2. LOX-Mediated ECM Stiffness Increases Cancer Cell Proliferation and Invasion
5. LOX Inhibitors
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Metastasis: Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Wu, M.; Wu, Z.-F.; Merajver, S.D. Rho proteins and cell-matrix interactions in cancer. Cells Tissues Organs 2007, 185, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM disrupts cancer progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef]
- Denton, A.E.; Roberts, E.W.; Fearon, D.T. Stromal Cells in the Tumor Microenvironment. In Stromal Immunology; Springer: Berlin, Germany, 2018; pp. 99–114. [Google Scholar]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, B.S.; Sternlicht, M.D.; Lund, L.R.; Alexander, C.M.; Mott, J.; Bissell, M.J.; Soloway, P.; Itohara, S.; Werb, Z. Site-specific inductive and inhibitory activities of MMP-2 and MMP-3 orchestrate mammary gland branching morphogenesis. J. Cell Biol. 2003, 162, 1123–1133. [Google Scholar] [CrossRef]
- Rebustini, I.T.; Myers, C.; Lassiter, K.S.; Surmak, A.; Szabova, L.; Holmbeck, K.; Pedchenko, V.; Hudson, B.G.; Hoffman, M.P. MT2-MMP-dependent release of collagen IV NC1 domains regulates submandibular gland branching morphogenesis. Dev. Cell 2009, 17, 482–493. [Google Scholar] [CrossRef]
- Jones, F.S.; Jones, P.L. The tenascin family of ECM glycoproteins: Structure, function, and regulation during embryonic development and tissue remodeling. Dev. Dyn. 2000, 218, 235–259. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Molecular pathways: Connecting fibrosis and solid tumor metastasis. Clin. Cancer Res. 2014, 20, 3637–3643. [Google Scholar] [CrossRef] [PubMed]
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 119–150. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Kenny, P.A.; Bissell, M.J. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via EMT? J. Cell. Biochem. 2007, 101, 830–839. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef]
- Zucker, S.; Vacirca, J. Role of matrix metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis Rev. 2004, 23, 101–117. [Google Scholar] [CrossRef]
- Casey, T.; Bond, J.; Tighe, S.; Hunter, T.; Lintault, L.; Patel, O.; Eneman, J.; Crocker, A.; White, J.; Tessitore, J. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res. Treat. 2009, 114, 47–62. [Google Scholar] [CrossRef]
- Crawford, N.P.; Walker, R.C.; Lukes, L.; Officewala, J.S.; Williams, R.W.; Hunter, K.W. The Diasporin Pathway: A tumor progression-related transcriptional network that predicts breast cancer survival. Clin. Exp. Metastasis 2008, 25, 357–369. [Google Scholar] [CrossRef]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108. [Google Scholar] [CrossRef]
- Sinkus, R.; Lorenzen, J.; Schrader, D.; Lorenzen, M.; Dargatz, M.; Holz, D. High-resolution tensor MR elastography for breast tumour detection. Phys. Med. Biol. 2000, 45, 1649. [Google Scholar] [CrossRef]
- Levental, K.R. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.; Liphardt, J.; Hwang, E.; Weaver, V. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.-M.; Wang, H.-B.; Dembo, M.; Wang, Y.-L. Cell movement is guided by the rigidity of the substrate. Biophys. J. 2000, 79, 144–152. [Google Scholar] [CrossRef]
- Wullkopf, L.; West, A.-K.V.; Leijnse, N.; Cox, T.R.; Madsen, C.D.; Oddershede, L.B.; Erler, J.T. Cancer cells’ ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Mol. Biol. Cell 2018, 29, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J. Metastatic growth from dormant cells induced by a col-I–enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Rumney, R.M.; Schoof, E.M.; Perryman, L.; Høye, A.M.; Agrawal, A.; Bird, D.; Ab Latif, N.; Forrest, H.; Evans, H.R. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 2015, 522, 106. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Yui, Y.; Damiano, L.; Bainer, R.O.; Lakins, J.N.; Acerbi, I.; Ou, G.; Wijekoon, A.C.; Levental, K.R.; Gilbert, P.M. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat. Med. 2014, 20, 360. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef]
- Cox, T.R.; Bird, D.; Baker, A.-M.; Barker, H.E.; Ho, M.W.; Lang, G.; Erler, J.T. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Koláčná, L.; Bakešová, J.; Varga, F.; Košťáková, E.; Planka, L.; Nečas, A.; Lukáš, D.; Amler, E.; Pelouch, V. Biochemical and biophysical aspects of collagen nanostructure in the extracellular matrix. Physiol. Res. 2007, 56, 51–60. [Google Scholar]
- Engler, A.J.; Griffin, M.A.; Sen, S.; Bönnemann, C.G.; Sweeney, H.L.; Discher, D.E. Myotubes differentiate optimally on substrates with tissue-like stiffness: Pathological implications for soft or stiff microenvironments. J. Cell Biol. 2004, 166, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Models Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef]
- Erler, J.T.; Weaver, V.M. Three-dimensional context regulation of metastasis. Clin. Exp. Metastasis 2009, 26, 35–49. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- van der Slot-Verhoeven, A.J.; van Dura, E.A.; Attema, J.; Blauw, B.; DeGroot, J.; Huizinga, T.W.; Zuurmond, A.-M.; Bank, R.A. The type of collagen cross-link determines the reversibility of experimental skin fibrosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2005, 1740, 60–67. [Google Scholar] [CrossRef]
- Colpaert, C.G.; Vermeulen, P.B.; Fox, S.B.; Harris, A.L.; Dirix, L.Y.; Van Marck, E.A. The presence of a fibrotic focus in invasive breast carcinoma correlates with the expression of carbonic anhydrase IX and is a marker of hypoxia and poor prognosis. Breast Cancer Res. Treat. 2003, 81, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, K. Lysyl oxidases: A novel multifunctional amine oxidase family. Prog. Nucleic Acid Res. Mol. Biol. 2001, 70, 1–32. [Google Scholar] [PubMed]
- Eyre, D.R.; Paz, M.A.; Gallop, P.M. Cross-linking in collagen and elastin. Annu. Rev. Biochem. 1984, 53, 717–748. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef]
- Mäki, J.M.; Räsänen, J.; Tikkanen, H.; Sormunen, R.; Mäkikallio, K.; Kivirikko, K.I.; Soininen, R. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation 2002, 106, 2503–2509. [Google Scholar] [CrossRef] [PubMed]
- Cronshaw, A.D.; Fothergill-Gilmore, L.A.; Hulmes, D. The proteolytic processing site of the precursor of lysyl oxidase. Biochem. J. 1995, 306, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Uzel, M.I.; Scott, I.C.; Babakhanlou-Chase, H.; Palamakumbura, A.H.; Pappano, W.N.; Hong, H.-H.; Greenspan, D.S.; Trackman, P.C. Multiple bone morphogenetic protein 1-related mammalian metalloproteinases process pro-lysyl oxidase at the correct physiological site and control lysyl oxidase activation in mouse embryo fibroblast cultures. J. Biol. Chem. 2001, 276, 22537–22543. [Google Scholar] [CrossRef]
- Grau-Bové, X.; Ruiz-Trillo, I.; Rodriguez-Pascual, F. Origin and evolution of lysyl oxidases. Sci. Rep. 2015, 5, 10568. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, Y.; Gao, J.; Pawlyk, B.; Starcher, B.; Spencer, J.A.; Yanagisawa, H.; Zuo, J.; Li, T. Elastic fiber homeostasis requires lysyl oxidase–like 1 protein. Nat. Genet. 2004, 36, 178. [Google Scholar] [CrossRef]
- Martínez, V.G.; Moestrup, S.K.; Holmskov, U.; Mollenhauer, J.; Lozano, F. The conserved scavenger receptor cysteine-rich superfamily in therapy and diagnosis. Pharmacol. Rev. 2011, 63, 967–1000. [Google Scholar] [CrossRef] [PubMed]
- Bignon, M.; Pichol-Thievend, C.; Hardouin, J.; Malbouyres, M.; Bréchot, N.; Nasciutti, L.; Barret, A.; Teillon, J.; Guillon, E.; Etienne, E. Lysyl oxidase-like protein-2 regulates sprouting angiogenesis and type IV collagen assembly in the endothelial basement membrane. Blood 2011, 18, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, O.; González-Santamaría, J.; Lagares, D.; Guinea-Viniegra, J.; Pichol-Thievend, C.; Muller, L.; Rodríguez-Pascual, F. LOXL4 is induced by TGF-β1 through Smad and JunB/Fra2 and contributes to vascular matrix remodeling. Mol. Cell. Biol. 2013, 33, 2388–2401. [Google Scholar] [CrossRef] [PubMed]
- Añazco, C.; López-Jiménez, A.J.; Rafi, M.; Vega-Montoto, L.; Zhang, M.-Z.; Hudson, B.G.; Vanacore, R.M. Lysyl Oxidase Like-2 Crosslinks Collagen IV of Glomerular Basement Membrane. J. Biol. Chem. 2016, 291, 25999–26012. [Google Scholar] [CrossRef]
- Schmelzer, C.E.; Heinz, A.; Troilo, H.; Lockhart-Cairns, M.P.; Jowitt, T.A.; Marchand, M.F.; Bidault, L.; Bignon, M.; Hedtke, T.; Barret, A. Lysyl oxidase–like 2 (LOXL2)–mediated cross-linking of tropoelastin. FASEB J. 2019. [Google Scholar] [CrossRef]
- Martin, A.; Salvador, F.; Moreno-Bueno, G.; Floristán, A.; Ruiz-Herguido, C.; Cuevas, E.P.; Morales, S.; Santos, V.; Csiszar, K.; Dubus, P. Lysyl oxidase-like 2 represses Notch1 expression in the skin to promote squamous cell carcinoma progression. EMBO J. 2015, 34, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, R.; Liu, Z.; Hou, C.; Zong, W.; Zhang, A.; Sun, X.; Gao, J. Loss of lysyl oxidase-like 3 causes cleft palate and spinal deformity in mice. Hum. Mol. Genet. 2015, 24, 6174–6185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Z.; Zhang, T.; Lin, Z.; Li, Z.; Zhang, A.; Sun, X.; Gao, J. Loss of lysyl oxidase-like 3 attenuates embryonic lung development in mice. Sci. Rep. 2016, 6, 33856. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Roh, S.; Park, J.-Y.; Kim, Y.; Cho, D.H.; Kim, J.C. Differential expression of the LOX family genes in human colorectal adenocarcinomas. Oncol. Rep. 2009, 22, 799–804. [Google Scholar] [CrossRef]
- Weise, J.B. LOXL4 is a selectively expressed candidate diagnostic antigen in head and neck cancer. Eur. J. Cancer 2008, 44, 1323–1331. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Q.; Wu, J.; Wang, J.; Shi, Y.; Liu, M. Crystal structure of human lysyl oxidase-like 2 (hLOXL2) in a precursor state. Proc. Natl. Acad. Sci. USA 2018, 115, 3828–3833. [Google Scholar] [CrossRef]
- Rucker, R.B.; Kosonen, T.; Clegg, M.S.; Mitchell, A.E.; Rucker, B.R.; Uriu-Hare, J.Y.; Keen, C.L. Copper, lysyl oxidase, and extracellular matrix protein cross-linking. Am. J. Clin. Nutr. 1998, 67 (Suppl. S5), 996S–1002S. [Google Scholar] [CrossRef]
- Shanbhag, V.; Jasmer-McDonald, K.; Zhu, S.; Martin, A.L.; Gudekar, N.; Khan, A.; Ladomersky, E.; Singh, K.; Weisman, G.A.; Petris, M.J. ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Opsahl, W.; Zeronian, H.; Ellison, M.; Lewis, D.; Rucker, R.B.; Riggins, R.S. Role of copper in collagen cross-linking and its influence on selected mechanical properties of chick bone and tendon. J. Nutr. 1982, 112, 708–716. [Google Scholar] [CrossRef]
- Tümer, Z.; Møller, L.B. Menkes disease. Eur. J. Hum. Genet. 2010, 18, 511. [Google Scholar] [CrossRef]
- Tümer, Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum. Mut. 2013, 34, 417–429. [Google Scholar] [CrossRef]
- Kuo, H.W.; Chen, S.F.; Wu, C.C.; Chen, D.R.; Lee, J.H. Serum and tissue trace elements in patients with breast cancer in Taiwan. Biol. Trace Elem. Res. 2002, 89, 1–11. [Google Scholar] [CrossRef]
- Rizk, S.L.; Sky-Peck, H.H. Comparison between concentrations of trace elements in normal and neoplastic human breast tissue. Cancer Res. 1984, 44, 5390–5394. [Google Scholar]
- Yaman, M.; Kaya, G.; Simsek, M. Comparison of trace element concentrations in cancerous and noncancerous human endometrial and ovary tissues. Int. J. Gynecol. Cancer 2007, 17, 220–228. [Google Scholar] [CrossRef]
- Santoliquido, P.M.; Southwick, H.W.; Olwin, J.H. Trace metal levels in cancer of the breast. Surg. Gynecol. Obstet. 1976, 142, 65–70. [Google Scholar]
- Geraki, K.; Farquharson, M.J.; Bradley, D.A. Concentrations of Fe, Cu and Zn in breast tissue: A synchrotron XRF study. Phys. Med. Biol. 2002, 47, 2327–2339. [Google Scholar] [CrossRef]
- Margalioth, E.J.; Schenker, J.G.; Chevion, M. Copper and zinc levels in normal and malignant tissues. Cancer 1983, 52, 868–872. [Google Scholar] [CrossRef]
- Mulay, I.L.; Roy, R.; Knox, B.E.; Suhr, N.H.; Delaney, W.E. Trace-metal analysis of cancerous and noncancerous human tissues. J. Natl. Cancer Inst. 1971, 47, 1–13. [Google Scholar]
- Lightman, A.; Brandes, J.M.; Binur, N.; Drugan, A.; Zinder, O. Use of the serum copper/zinc ratio in the differential diagnosis of ovarian malignancy. Clin. Chem. 1986, 32, 101–103. [Google Scholar]
- Zimnicka, A.M.; Tang, H.; Guo, Q.; Kuhr, F.K.; Oh, M.J.; Wan, J.; Chen, J.; Smith, K.A.; Fraidenburg, D.R.; Choudhury, M.S.; Levitan, I.; Machado, R.F.; Kaplan, J.H.; Yuan, J.X. Upregulated copper transporters in hypoxia-induced pulmonary hypertension. PLoS ONE 2014, 9, e90544. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.L.; Hendrix, M.J.; Kirschmann, D.A. Paradoxical roles for lysyl oxidases in cancer—A prospect. J. Cell. Biochem. 2007, 101, 1338–1354. [Google Scholar] [CrossRef]
- Kirschmann, D.A. A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res. 2002, 62, 4478–4483. [Google Scholar]
- Salvador, F.; Martin, A.; López-Menéndez, C.; Moreno-Bueno, G.; Santos, V.; Vázquez-Naharro, A.; Santamaria, P.G.; Morales, S.; Dubus, P.R.; Muinelo-Romay, L. Lysyl Oxidase–like Protein LOXL2 Promotes Lung Metastasis of Breast Cancer. Cancer Res. 2017, 77, 5846–5859. [Google Scholar] [CrossRef]
- Baker, A.; Bird, D.; Lang, G.; Cox, T.R.; Erler, J. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene 2013, 32, 1863. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.-M.; Cox, T.R.; Bird, D.; Lang, G.; Murray, G.I.; Sun, X.-F.; Southall, S.M.; Wilson, J.R.; Erler, J.T. The role of lysyl oxidase in SRC-dependent proliferation and metastasis of colorectal cancer. J. Natl. Cancer Inst. 2011, 103, 407–424. [Google Scholar] [CrossRef]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: Inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Li, C.; Higgins, J.P.; Van De Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef]
- Cheon, D.-J.; Tong, Y.; Sim, M.-S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E. A collagen-remodeling gene signature regulated by TGF-β signaling is associated with metastasis and poor survival in serous ovarian cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef]
- Ryner, L.; Guan, Y.; Firestein, R.; Xiao, Y.; Choi, Y.; Rabe, C.; Lu, S.; Fuentes, E.; Huw, L.-Y.; Lackner, M.R. Upregulation of periostin and reactive stroma is associated with primary chemoresistance and predicts clinical outcomes in epithelial ovarian cancer. Clin. Cancer Res. 2015, 21, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.-T.; Harris, J.; Magliocco, A.M.; Kong, C.S.; Diaz, R.; Shin, B.; Cao, H.; Trotti, A.; Erler, J.T.; Chung, C.H. Validation of lysyl oxidase as a prognostic marker for metastasis and survival in head and neck squamous cell carcinoma: Radiation Therapy Oncology Group trial 90-03. J. Clin. Oncol. 2009, 27, 4281. [Google Scholar] [CrossRef] [PubMed]
- Gorogh, T. Selective upregulation and amplification of the lysyl oxidase like-4 (LOXL4) gene in head and neck squamous cell carcinoma. J. Pathol. 2007, 212, 74–82. [Google Scholar] [CrossRef]
- Albinger-Hegyi, A.; Stoeckli, S.J.; Schmid, S.; Storz, M.; Iotzova, G.; Probst-Hensch, N.M.; Rehrauer, H.; Tinguely, M.; Moch, H.; Hegyi, I. Lysyl oxidase expression is an independent marker of prognosis and a predictor of lymph node metastasis in oral and oropharyngeal squamous cell carcinoma (OSCC). Int. J. Cancer 2010, 126, 2653–2662. [Google Scholar] [CrossRef] [PubMed]
- Hase, H.; Jingushi, K.; Ueda, Y.; Kitae, K.; Egawa, H.; Ohshio, I.; Kawakami, R.; Kashiwagi, Y.; Tsukada, Y.; Kobayashi, T. LOXL2 status correlates with tumor stage and regulates integrin levels to promote tumor progression in ccRCC. Mol. Cancer Res. 2014, 12, 1807–1817. [Google Scholar] [CrossRef]
- Abourbih, D.A.; Di Cesare, S.; Orellana, M.E.; Antecka, E.; Martins, C.; Petruccelli, L.A.; Burnier Jr, M.N. Lysyl oxidase expression and inhibition in uveal melanoma. Melanoma Res. 2010, 20, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.K.; Csiszar, K. Intracellular localization of the matrix enzyme lysyl oxidase in polarized epithelial cells. Matrix Biol. 2007, 26, 136–139. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Payne, S.L. Lysyl oxidase regulates breast cancer cell migration and adhesion through a hydrogen peroxide-mediated mechanism. Cancer Res. 2005, 65, 11429–11436. [Google Scholar] [CrossRef]
- Peinado, H. A molecular role for lysyl oxidase-like 2 enzyme in snail regulation and tumor progression. EMBO J. 2005, 24, 3446–3458. [Google Scholar] [CrossRef]
- Gao, Y.; Xiao, Q.; Ma, H.; Li, L.; Liu, J.; Feng, Y.; Fang, Z.; Wu, J.; Han, X.; Zhang, J. LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proc. Natl. Acad. Sci. USA 2010, 107, 18892–18897. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.; Foreman, K.M.; Soriano, M.I.; Rossen, N.S.; Shehade, H.; Fregoso, D.R.; Eggold, J.T.; Krishnan, V.; Dorigo, O.; Krieg, A.J. Collagen remodeling in the hypoxic tumor-mesothelial niche promotes ovarian cancer metastasis. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H. Lysyl oxidase-like 2 as a new poor prognosis marker of squamous cell carcinomas. Cancer Res. 2008, 68, 4541–4550. [Google Scholar] [CrossRef]
- Grutzmann, R. Gene expression profiles of microdissected pancreatic ductal adenocarcinoma. Virchows Arch. 2003, 443, 508–517. [Google Scholar] [CrossRef]
- Barker, H.E.; Chang, J.; Cox, T.R.; Lang, G.; Bird, D.; Nicolau, M.; Evans, H.R.; Gartland, A.; Erler, J.T. LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res. 2011, 71, 1561–1572. [Google Scholar] [CrossRef]
- Akiri, G. Lysyl oxidase-related protein-1 promotes tumor fibrosis and tumor progression in vivo. Cancer Res. 2003, 63, 1657–1666. [Google Scholar]
- Ahn, S.G.; Dong, S.M.; Oshima, A.; Kim, W.H.; Lee, H.M.; Lee, S.A.; Kwon, S.-h.; Lee, J.-h.; Lee, J.M.; Jeong, J. LOXL2 expression is associated with invasiveness and negatively influences survival in breast cancer patients. Breast Cancer Res. Treat. 2013, 141, 89–99. [Google Scholar] [CrossRef]
- Moreno-Bueno, G. Lysyl oxidase-like 2 (LOXL2), a new regulator of cell polarity required for metastatic dissemination of basal-like breast carcinomas. EMBO Mol. Med. 2011, 3, 528–544. [Google Scholar] [CrossRef]
- Fong, S.F. Lysyl oxidase-like 2 expression is increased in colon and esophageal tumors and associated with less differentiated colon tumors. Genes Chromosom Cancer 2007, 46, 644–655. [Google Scholar] [CrossRef]
- Peng, L. Secreted LOXL2 is a novel therapeutic target that promotes gastric cancer metastasis via the Src/FAK pathway. Carcinogenesis 2009, 30, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Garcia-Palmero, I.; Herrera, M.; Bartolomé, R.A.; Peña, C.; Fernandez-Aceñero, M.J.; Padilla, G.; Peláez-García, A.; Lopez-Lucendo, M.; Rodriguez-Merlo, R. LOXL2 is highly expressed in cancer-associated fibroblasts and associates to poor colon cancer survival. Clin. Cancer Res. 2015, 21, 4892–4902. [Google Scholar] [CrossRef]
- Barry-Hamilton, V. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 2010, 16, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Li, R.-k.; Zhao, W.-y.; Fang, F.; Zhuang, C.; Zhang, X.-x.; Yang, X.-m.; Jiang, S.-h.; Kong, F.-z.; Tu, L.; Zhang, W.-M. Lysyl oxidase-like 4 (LOXL4) promotes proliferation and metastasis of gastric cancer via FAK/Src pathway. J. Cancer Res. Clin. Oncol. 2015, 141, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Kim, H.S.; Jin, T.; Moon, W.K. LOXL4 knockdown enhances tumor growth and lung metastasis through collagen-dependent extracellular matrix changes in triple-negative breast cancer. Oncotarget 2017, 8, 11977. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Umesh, V.; Rape, A.D.; Ulrich, T.A.; Kumar, S. Microenvironmental stiffness enhances glioma cell proliferation by stimulating epidermal growth factor receptor signaling. PLoS ONE 2014, 9, e101771. [Google Scholar] [CrossRef] [PubMed]
- Solon, J.; Levental, I.; Sengupta, K.; Georges, P.C.; Janmey, P.A. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys. J. 2007, 93, 4453–4461. [Google Scholar] [CrossRef]
- Murray, P.; Frampton, G.; Nelson, P.N. Cell adhesion molecules. Sticky moments in the clinic. BMJ 1999, 319, 332–334. [Google Scholar] [CrossRef]
- Horton, E.R.; Byron, A.; Askari, J.A.; Ng, D.H.J.; Millon-Fremillon, A.; Robertson, J.; Koper, E.J.; Paul, N.R.; Warwood, S.; Knight, D.; Humphries, J.D.; Humphries, M.J. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat. Cell Biol. 2015, 17, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zheng, Q.; Xing, X.; Dong, Y.; Wang, Y.; You, Y.; Chen, R.; Hu, C.; Chen, J.; Gao, D. Matrix stiffness-upregulated LOXL2 promotes fibronectin production, MMP9 and CXCL12 expression and BMDCs recruitment to assist pre-metastatic niche formation. J. Exp. Clin. Cancer Res. 2018, 37, 99. [Google Scholar] [CrossRef]
- Dong, Y.; Xie, X.; Wang, Z.; Hu, C.; Zheng, Q.; Wang, Y.; Chen, R.; Xue, T.; Chen, J.; Gao, D. Increasing matrix stiffness upregulates vascular endothelial growth factor expression in hepatocellular carcinoma cells mediated by integrin β1. Biochem. Biophys. Res. Commun. 2014, 444, 427–432. [Google Scholar] [CrossRef]
- Gao, A.E.; Sullivan, K.E.; Black, L.D., III. Lysyl oxidase expression in cardiac fibroblasts is regulated by α2β1 integrin interactions with the cellular microenvironment. Biochem. Biophys. Res. Commun. 2016, 475, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Voloshenyuk, T.G.; Landesman, E.S.; Khoutorova, E.; Hart, A.D.; Gardner, J.D. Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine 2011, 55, 90–97. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 1. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.N.; Jeibmann, A.; Halama, K.; Witte, H.T.; Wälte, M.; Matzat, T.; Schillers, H.; Faber, C.; Senner, V.; Paulus, W. ECM stiffness regulates glial migration in Drosophila and mammalian glioma models. Development 2014, 141, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Navab, R.; Strumpf, D.; To, C.; Pasko, E.; Kim, K.; Park, C.; Hai, J.; Liu, J.; Jonkman, J.; Barczyk, M. Integrin α11β1 regulates cancer stromal stiffness and promotes tumorigenicity and metastasis in non-small cell lung cancer. Oncogene 2016, 35, 1899. [Google Scholar] [CrossRef]
- Paszek, M.J.; Weaver, V.M. The tension mounts: Mechanics meets morphogenesis and malignancy. J. Mammary Gland Biol. Neoplasia 2004, 9, 325–342. [Google Scholar] [CrossRef]
- Du, J.; Chen, X.; Liang, X.; Zhang, G.; Xu, J.; He, L.; Zhan, Q.; Feng, X.Q.; Chien, S.; Yang, C. Integrin activation and internalization on soft ECM as a mechanism of induction of stem cell differentiation by ECM elasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 9466–9471. [Google Scholar] [CrossRef]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816. [Google Scholar] [CrossRef]
- Cassereau, L.; Miroshnikova, Y.A.; Ou, G.; Lakins, J.; Weaver, V.M. A 3D tension bioreactor platform to study the interplay between ECM stiffness and tumor phenotype. J. Biotechnol. 2015, 193, 66–69. [Google Scholar] [CrossRef]
- Miroshnikova, Y.; Jorgens, D.; Spirio, L.; Auer, M.; Sarang-Sieminski, A.; Weaver, V. Engineering strategies to recapitulate epithelial morphogenesis within synthetic three-dimensional extracellular matrix with tunable mechanical properties. Phys. Biol. 2011, 8, 026013. [Google Scholar] [CrossRef]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK–ERK linkage. Oncogene 2009, 28, 4326. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Desai, R.; Solski, P.A.; Der, C.J.; Keely, P.J. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J. Cell Biol. 2003, 163, 583–595. [Google Scholar] [CrossRef]
- Wyckoff, J.B.; Pinner, S.E.; Gschmeissner, S.; Condeelis, J.S.; Sahai, E. ROCK-and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr. Biol. 2006, 16, 1515–1523. [Google Scholar] [CrossRef]
- Lahlou, H.; Sanguin-Gendreau, V.; Zuo, D.; Cardiff, R.D.; McLean, G.W.; Frame, M.C.; Muller, W.J. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc. Natl. Acad. Sci. USA 2007, 104, 20302–20307. [Google Scholar] [CrossRef] [PubMed]
- White, D.E.; Kurpios, N.A.; Zuo, D.; Hassell, J.A.; Blaess, S.; Mueller, U.; Muller, W.J. Targeted disruption of β1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell 2004, 6, 159–170. [Google Scholar] [CrossRef]
- Pelham, R.J.; Wang, Y.-L. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 1997, 94, 13661–13665. [Google Scholar] [CrossRef]
- Artym, V.V.; Swatkoski, S.; Matsumoto, K.; Campbell, C.B.; Petrie, R.J.; Dimitriadis, E.K.; Li, X.; Mueller, S.C.; Bugge, T.H.; Gucek, M.; Yamada, K.M. Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network. J. Cell Biol. 2015, 208, 331–350. [Google Scholar] [CrossRef] [PubMed]
- Bondareva, A.; Downey, C.M.; Ayres, F.; Liu, W.; Boyd, S.K.; Hallgrimsson, B.; Jirik, F.R. The lysyl oxidase inhibitor, β-aminopropionitrile, diminishes the metastatic colonization potential of circulating breast cancer cells. PLoS ONE 2009, 4, e5620. [Google Scholar] [CrossRef]
- Pickup, M.W.; Laklai, H.; Acerbi, I.; Owens, P.; Gorska, A.E.; Chytil, A.; Aakre, M.; Weaver, V.M.; Moses, H.L. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-β–deficient mouse mammary carcinomas. Cancer Res. 2013, 73, 5336–5346. [Google Scholar] [CrossRef]
- Barker, H.E.; Bird, D.; Lang, G.; Erler, J.T. Tumor-secreted LOXL2 activates fibroblasts through FAK signaling. Mol. Cancer Res. 2013, 11, 1425–1436. [Google Scholar] [CrossRef]
- Jung, S.T.; Kim, M.S.; Seo, J.Y.; Kim, H.C.; Kim, Y. Purification of enzymatically active human lysyl oxidase and lysyl oxidase-like protein from Escherichia coli inclusion bodies. Protein Exp. Purif. 2003, 31, 240–246. [Google Scholar] [CrossRef]
- Kim, M.S. Expression and purification of enzymatically active forms of the human lysyl oxidase-like protein 4. J. Biol. Chem. 2003, 278, 52071–52074. [Google Scholar] [CrossRef] [PubMed]
- Hajdú, I.; Kardos, J.; Major, B.; Fabó, G.; Lőrincz, Z.; Cseh, S.; Dormán, G. Inhibition of the LOX enzyme family members with old and new ligands. Selectivity analysis revisited. Bioorg. Med. Chem. Lett. 2018, 28, 3113–3118. [Google Scholar] [CrossRef]
- Tang, S.S.; Simpson, D.; Kagan, H. Beta-substituted ethylamine derivatives as suicide inhibitors of lysyl oxidase. J. Biol. Chem. 1984, 259, 975–979. [Google Scholar]
- Liu, S.B.; Ikenaga, N.; Peng, Z.-W.; Sverdlov, D.Y.; Greenstein, A.; Smith, V.; Schuppan, D.; Popov, Y. Lysyl oxidase activity contributes to collagen stabilization during liver fibrosis progression and limits spontaneous fibrosis reversal in mice. FASEB J. 2015, 30, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Keiser, H.R.; Sjoerdsma, A. Studies on beta-aminopropionitrile in patients with scleroderma. Clin. Pharmacol. Ther. 1967, 8, 593–602. [Google Scholar] [CrossRef]
- Peacock, E.E.; Madden, J.W. Some studies on the effects of β-aminopropionitrile in patients with injured flexor tendons. Surgery 1969, 66, 215–223. [Google Scholar]
- Chvapil, M.; Misiorowski, R.; Eskelson, C. On the mechanisms of β-aminopropionitrile toxicity. J. Surg. Res. 1981, 31, 151–155. [Google Scholar] [CrossRef]
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology 2018, 155, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Brown, K.K.; Collard, H.R.; Cottin, V.; Gibson, K.F.; Kaner, R.J.; Lederer, D.J.; Martinez, F.J.; Noble, P.W.; Song, J.W. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: A randomised, double-blind, controlled, phase 2 trial. Lancet Respir. Med. 2017, 5, 22–32. [Google Scholar] [CrossRef]
- Spagnolo, P.; Maher, T.M. Clinical trial research in focus: Why do so many clinical trials fail in IPF? Lancet Respir. Med. 2017, 5, 372–374. [Google Scholar] [CrossRef]
- Puente, A.; Fortea, J.I.; Cabezas, J.; Arias Loste, M.T.; Iruzubieta, P.; Llerena, S.; Huelin, P.; Fábrega, E.; Crespo, J. LOXL2—A New Target in Antifibrogenic Therapy? Int. J. Mol. Sci. 2019, 20, 1634. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.H.; Rowbottom, M.W.; Lonergan, D.; Darlington, J.; Prodanovich, P.; King, C.D.; Evans, J.F.; Bain, G. Small molecule lysyl oxidase-like 2 (LOXL2) inhibitors: The identification of an inhibitor selective for LOXL2 over LOX. ACS Med. Chem. Lett. 2017, 8, 423–427. [Google Scholar] [CrossRef]
- Rowbottom, M.W.; Bain, G.; Calderon, I.; Lasof, T.; Lonergan, D.; Lai, A.; Huang, F.; Darlington, J.; Prodanovich, P.; Santini, A.M. Identification of 4-(aminomethyl)-6-(trifluoromethyl)-2-(phenoxy) pyridine derivatives as potent, selective, and orally efficacious inhibitors of the copper-dependent amine oxidase, lysyl oxidase-like 2 (LOXL2). J. Med. Chem. 2017, 60, 4403–4423. [Google Scholar] [CrossRef]
- Findlay, A.D.; Turner, C.I.; Deodhar, M.; Foot, J.S.; Jarolimek, W.; Zhou, W.; Robertson, A.D. Indole and azaindole haloallylamine derivative inhibitors of lysyl oxidases and uses thereof. WO2017136871A1. 17 August 2017. [Google Scholar]
- Schilter, H.; Findlay, A.D.; Perryman, L.; Yow, T.T.; Moses, J.; Zahoor, A.; Turner, C.I.; Deodhar, M.; Foot, J.S.; Zhou, W. The lysyl oxidase like 2/3 enzymatic inhibitor, PXS-5153A, reduces crosslinks and ameliorates fibrosis. J. Cell. Mol. Med. 2019, 23, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.; Toshniwal, P.; Clemons, T.; Stevenson, A.; Ryan, E.; Jarolimek, W.; Wood, F.; Fear, M. 527 Targeting Lysyl Oxidase (LOX) Activity to Improve Scar Appearance. J. Burn Care Res. 2018, 39 (Suppl. S1), S237. [Google Scholar] [CrossRef]
- Chang, J.; Lucas, M.C.; Leonte, L.E.; Garcia-Montolio, M.; Singh, L.B.; Findlay, A.D.; Deodhar, M.; Foot, J.S.; Jarolimek, W.; Timpson, P. Pre-clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 2017, 8, 26066. [Google Scholar] [CrossRef]
- Tang, H.; Leung, L.; Saturno, G.; Viros, A.; Smith, D.; Di Leva, G.; Morrison, E.; Niculescu-Duvaz, D.; Lopes, F.; Johnson, L. Lysyl oxidase drives tumour progression by trapping EGF receptors at the cell surface. Nat. Commun. 2017, 8, 14909. [Google Scholar] [CrossRef]
- Discher, D.E.; Janmey, P.; Wang, Y.L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G. The role of matrix stiffness in regulating cell behavior. Hepatology 2008, 47, 1394–1400. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amendola, P.G.; Reuten, R.; Erler, J.T. Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment. Cancers 2019, 11, 729. https://doi.org/10.3390/cancers11050729
Amendola PG, Reuten R, Erler JT. Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment. Cancers. 2019; 11(5):729. https://doi.org/10.3390/cancers11050729
Chicago/Turabian StyleAmendola, Pier Giorgio, Raphael Reuten, and Janine Terra Erler. 2019. "Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment" Cancers 11, no. 5: 729. https://doi.org/10.3390/cancers11050729
APA StyleAmendola, P. G., Reuten, R., & Erler, J. T. (2019). Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment. Cancers, 11(5), 729. https://doi.org/10.3390/cancers11050729