Epigenetic Reprogramming of TGF-β Signaling in Breast Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Histone Modifications Govern Access of Transcription Factors to DNA
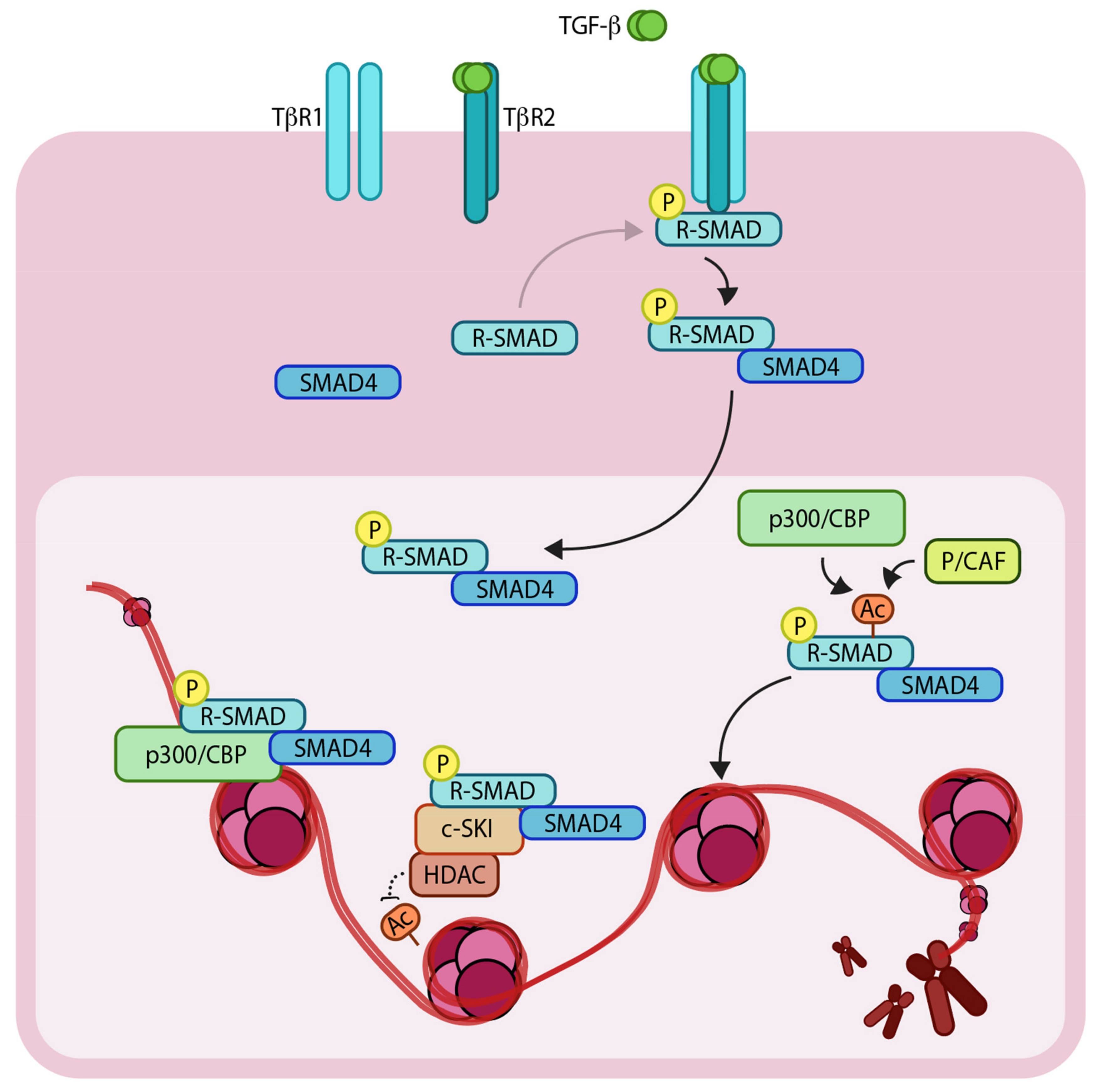
1.2. Epigenetic Regulators Modify TGF-β Signaling Components to Control the Genetic Output
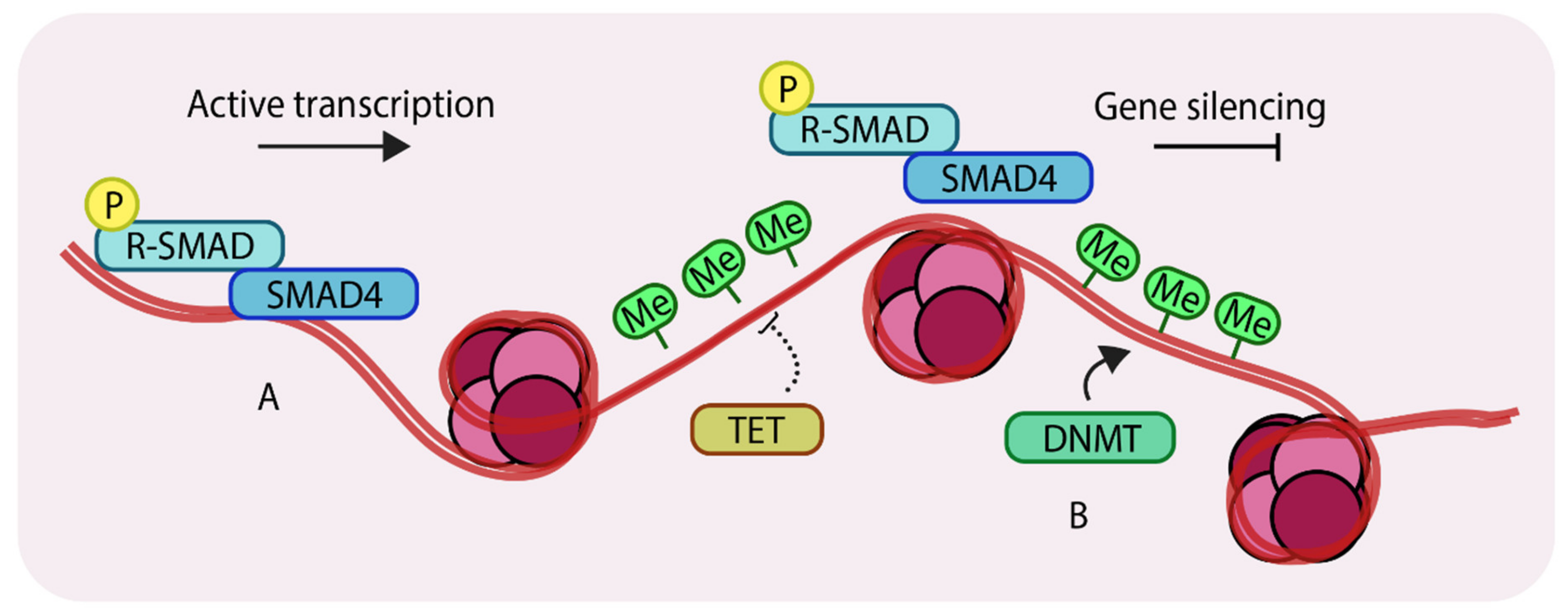
1.3. Epigenetic Changes that Take Place at the Genomic DNA Level
2. Role of DNA Methylation in Breast Development and Tumorigenesis
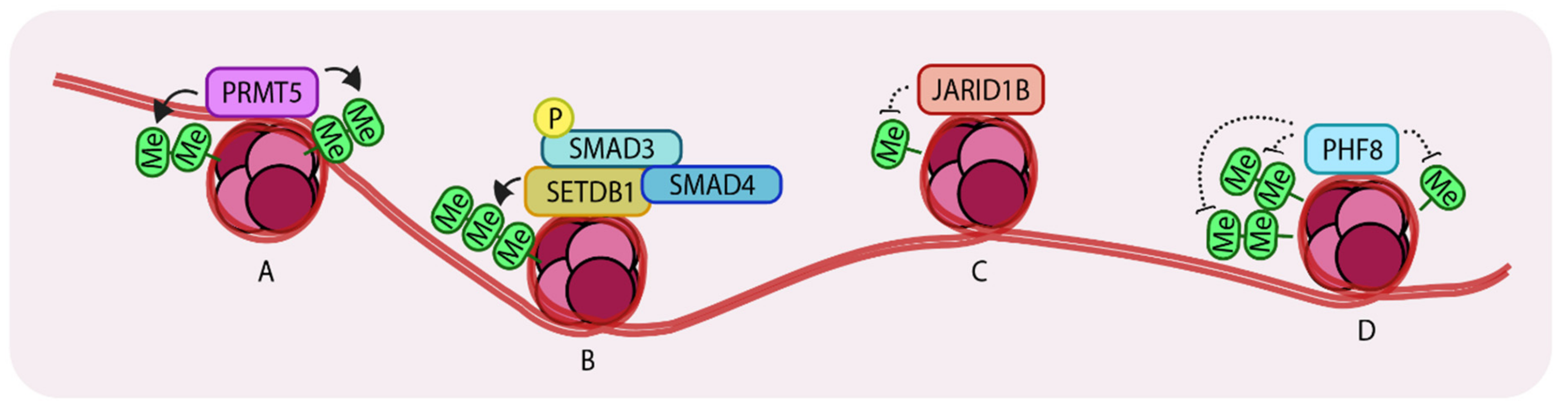
3. Methylation Status of Histones Govern TGF-β Mediated Changes
3.1. PRMT5 Augments TGF-β-Mediated EMT
3.2. An Interplay between Acetylation and Methylation by SETDB1
3.3. JARID1B Controls TGF-β-Mediated Growth Arrest
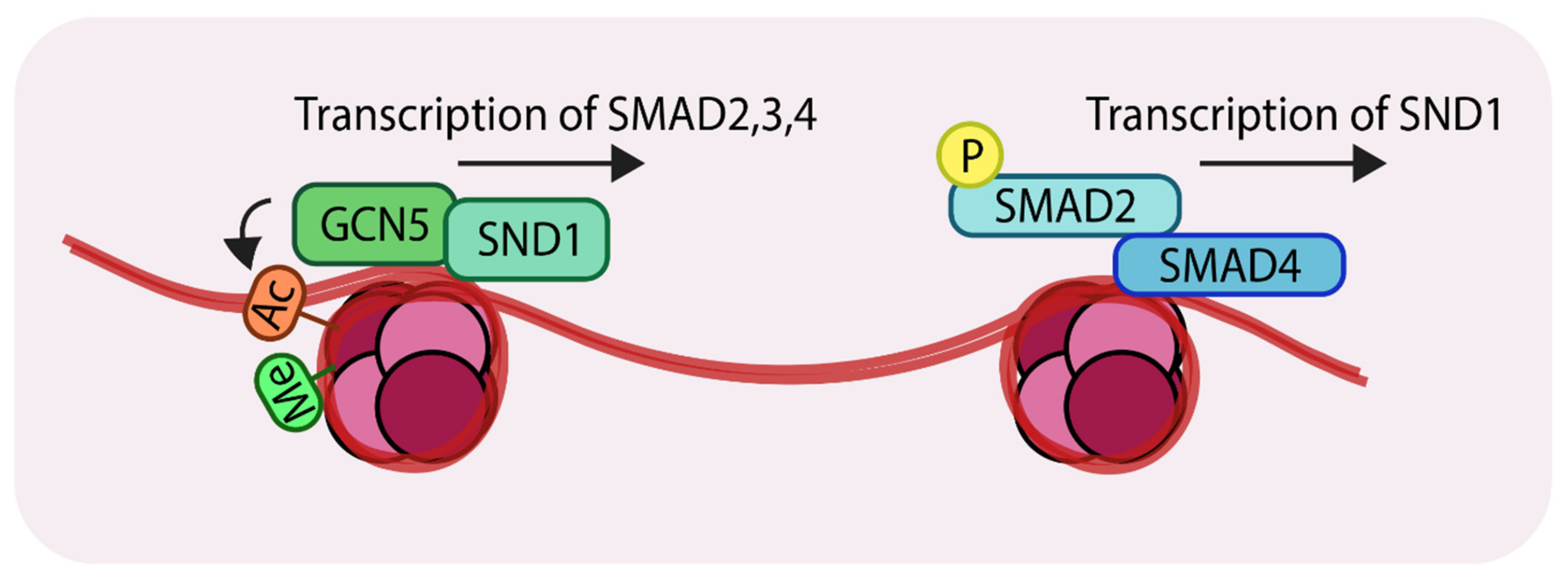
3.4. A Subunit of the LSD1-CoREST Complex Controls the Expression of SNAIL
3.5. KDM6B Stimulates SNAI1 Expression by Removing H3K27me3
3.6. Demethylation by PHF8 Enhances EMT
4. An Interplay of Histone Acetylation and Deacetylation Regulates TGF-β Mediated Genetic Output
5. Emerging Epigenetic Roles of Non-Coding RNAs
6. Conclusions and Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| CBP | CREB-binding protein |
| ChIP-seq | Chromatin Immunoprecipitation-sequencing |
| c-SKI | Cytoplasmic-Sloan Kettering Institute |
| DNMT | DNA methyltransferase |
| GCN5 | General control non-repressed protein 5 |
| EMT | Epithelial to mesenchymal transition |
| ER+ | Estrogen receptor positive |
| H3 | Histone 3 |
| H4 | Histone 4 |
| H3K4 | Histone 3 lysine 4 |
| H3K9 | Histone 3 lysine 9 |
| H3K4Me3 | Histone 3 Lysine 4 tri-methylation |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| HMG20 | High mobility group domain containing protein 20 |
| HMLE | human mammary epithelial cells |
| HOTAIR | HOX transcript antisense RNA |
| JARID1B | Jumonji/ARID domain-containing protein 1B |
| LBH | Limb bud and heart development |
| LncATB | Long non-coding RNA, Activated by TGF-β |
| LncRNA | Long non-coding RNA |
| LSD1-CoREST | Lysine-specific demethylase 1/REST co-repressor 1 |
| MALAT | Metastasis associated lung adenocarcinoma transcript |
| MEP50 | Methylosome protein 50 |
| METTL14 | Methyltransferase like 14 |
| miRNA | microRNA |
| MMP9 | Matrix metallopeptidase 9 |
| Nanog | Derived from Tìr nan Òg, the mythical Celtic land of youth |
| p/CAF | p300/CBP associated factor |
| PHF8 | PHD finger protein 8 |
| PRMT | Protein arginine methyltransferase |
| SETDB1 | Set domain bifurcated 1 |
| SMAD | SMA and MAD related protein |
| SMURF2 | SMAD ubiquitination regulatory factor 2 |
| SNAI1 | snail family transcriptional repressor 1 |
| SND1 | Staphylococcal nuclease domain-containing 1 |
| SnoN | SKI-related novel protein N |
| SUV39H1/2 | Suppressor of variegation 3-9 homolog 1/2 |
| TCGA | The cancer genomic atlas |
| TET | Ten-eleven translocation |
| TGF-β | Transforming growth factor-β |
| TRIM33 | Tripartite motif-containing 33 |
| TSS | Transcription start site |
| TWIST1 | Twist family bHLH transcription factor 1 |
| TβR | TGF-β receptor |
| VEGF | Vascular endothelial growth factor |
| WTAP | Wilm’s tumor-1 associated protein |
| ZEB | Zinc finger E-box binding homeobox |
References
- Kang, J.S.; Liu, C.; Derynck, R. New regulatory mechanisms of TGF-β receptor function. Trends Cell Biol. 2009, 19, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming Growth Factor-β Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [PubMed]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Souchelnytskyi, S.; Heldin, C.H. Smad regulation in TGF-β signal transduction. J. Cell Sci. 2001, 114, 4359–4369. [Google Scholar] [PubMed]
- Zhang, Y.; Feng, X.; We, R.; Derynck, R. Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature 1996, 383, 168–172. [Google Scholar] [CrossRef]
- Lagna, G.; Hata, A.; Hemmati-Brivanlou, A.; Massagué, J. Partnership between DPC4 and SMAD proteins in TGF-β signalling pathways. Nature 1996, 383, 832–836. [Google Scholar] [CrossRef]
- David, C.J.; Massague, J. Contextual determinants of TGF-β action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF-β receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Drabsch, Y.; Ten Dijke, P. TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012, 31, 553–568. [Google Scholar] [CrossRef]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.M.S.; Moskaluk, C.A.; Da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, A Candidate Tumor Suppressor Gene at Human Chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Microbiol. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Plath, K.; Fang, J.; Mlynarczyk-Evans, S.K.; Cao, R.; Worringer, K.A.; Wang, H.; De La Cruz, C.C.; Otte, A.P.; Panning, B.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in X Inactivation. Science 2003, 300, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Murn, J.; Shi, Y. The winding path of protein methylation research: Milestones and new frontiers. Nat. Rev. Mol. Cell Biol. 2017, 18, 517–527. [Google Scholar] [CrossRef]
- Hassig, C.A.; Schreiber, S.L. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Chem. Biol. 1997, 1, 300–308. [Google Scholar] [CrossRef]
- Janknecht, R.; Wells, N.J.; Hunter, T. TGF-β-stimulated cooperation of Smad proteins with the coactivators CBP/p300. Genes Dev. 1998, 12, 2114–2119. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, M.; Kanduri, M.; Grönroos, E.; Heldin, C.-H.; Ericsson, J. The DNA Binding Activities of Smad2 and Smad3 Are Regulated by Coactivator-mediated Acetylation. J. Biol. Chem. 2006, 281, 39870–39880. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, M.; Ericsson, J.; Grönroos, E.; Heldin, C.-H. The Balance between acetylation and deacetylation Controls Smad7 Stability. J. Biol. Chem. 2005, 280, 21797–21803. [Google Scholar] [CrossRef]
- Katsuno, Y.; Qin, J.; Oses-Prieto, J.; Wang, H.; Jackson-Weaver, O.; Zhang, T.; Lamouille, S.; Wu, J.; Burlingame, A.; Xu, J.; et al. Arginine methylation of SMAD7 by PRMT1 in TGF-β-induced epithelial-mesenchymal transition and epithelial stem-cell generation. J. Biol. Chem. 2018, 293, 13059–13072. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, S.; Inoue, H.; Hanai, J.; Kusanagi, K.; Nemoto, N.; Miyazono, K.; Kawabata, M. c-Ski acts as a transcriptional co-repressor in transforming growth factor-β signaling through interaction with smads. J. Biol. Chem. 1999, 274, 35269–35277. [Google Scholar] [CrossRef] [PubMed]
- Stroschein, S.L.; Wang, W.; Zhou, S.; Zhou, Q.; Luo, K. Negative feedback regulation of TGF-β signaling by the SnoN oncoprotein. Science 1999, 286, 771–774. [Google Scholar] [CrossRef]
- Zhang, F.; Lundin, M.; Ristimäki, A.; Heikkilä, P.; Lundin, J.; Isola, J.; Joensuu, H.; Laiho, M. Ski-related novel protein N (SnoN), a negative controller of transforming growth factor-β signaling, is a prognostic marker in estrogen receptor-positive breast carcinomas. Cancer Res. 2003, 63, 5005–5010. [Google Scholar]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a deubiquitinating enzyme essential for TGF-β signaling, controls Smad4 monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Mihaylova, V.T.; Kuruvilla, L.; Chen, F.; Viviano, S.; Baldassarre, M.; Sperandio, D.; Martinez, R.; Yue, P.; Bates, J.G.; et al. Loss of TRIM33 causes resistance to BET bromodomain inhibitors through MYC- and TGF-β-dependent mechanisms. Proc. Natl. Acad. Sci. USA 2016, 113, 4558–4566. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Bird, A.P. CpG islands-‘a rough guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Microbiol. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef]
- Lamprecht, S.; Sigal-Batikoff, I.; Shany, S.; Abu-Freha, N.; Ling, E.; Delinasios, G.J.; Moyal-Atias, K.; Delinasios, J.G.; Fich, A. Teaming Up for Trouble: Cancer Cells, Transforming Growth Factor-β1 Signaling and the Epigenetic Corruption of Stromal Naive Fibroblasts. Cancers 2018, 10, 61. [Google Scholar] [CrossRef]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef]
- Iizuka-Koga, M.; Nakatsukasa, H.; Ito, M.; Akanuma, T.; Lu, Q.; Yoshimura, A. Induction and maintenance of regulatory T cells by transcription factors and epigenetic modifications. J. Autoimmun. 2017, 83, 113–121. [Google Scholar] [CrossRef]
- Ganesh, K.; Massague, J. TGF-β Inhibition and Immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Microbiol. 2012, 13, 679–692. [Google Scholar] [CrossRef]
- Hennighausen, L.; Robinson, G.W. Information networks in the mammary gland. Nat. Rev. Mol. Cell Biol. 2005, 6, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Pierce, D.F., Jr.; Johnson, M.D.; Matsui, Y.; Robinson, S.D.; Gold, L.I.; Purchio, A.F.; Daniel, C.W.; Hogan, B.L.; Moses, H.L. Inhibition of mammary duct development but not alveolar outgrowth during pregnancy in transgenic mice expressing active TGF-β 1. Genes Dev. 1993, 7, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Kordon, E.C.; McKnight, R.A.; Jhappan, C.; Hennighausen, L.; Merlino, G.; Smith, G.H. Ectopic TGF β 1 expression in the secretory mammary epithelium induces early senescence of the epithelial stem cell population. Dev. Biol. 1995, 168, 47–61. [Google Scholar] [CrossRef]
- Huh, S.J.; Clement, K.; Jee, D.; Merlini, A.; Choudhury, S.; Maruyama, R.; Yoo, R.; Chytil, A.; Boyle, P.; Ran, F.A.; et al. Age- and Pregnancy-Associated DNA Methylation Changes in Mammary Epithelial Cells. Stem Cell Rep. 2015, 4, 297–311. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Ruzinova, M.B.; Benezra, R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003, 13, 410–418. [Google Scholar] [CrossRef]
- Choudhury, S.; Almendro, V.; Merino, V.F.; Wu, Z.; Maruyama, R.; Su, Y.; Martins, F.C.; Fackler, M.J.; Bessarabova, M.; Kowalczyk, A.; et al. Molecular profiling of human mammary gland links breast cancer risk to a p27(+) cell population with progenitor characteristics. Cell Stem Cell 2013, 13, 117–130. [Google Scholar] [CrossRef]
- Tufegdzic Vidakovic, A.; Rueda, O.M.; Vervoort, S.J.; Sati Batra, A.; Goldgraben, M.A.; Uribe-Lewis, S.; Greenwood, W.; Coffer, P.J.; Bruna, A.; Caldas, C. Context-Specific Effects of TGF-β/SMAD3 in Cancer Are Modulated by the Epigenome. Cell Rep. 2015, 13, 2480–2490. [Google Scholar] [CrossRef]
- Bruna, A.; Greenwood, W.; Le Quesne, J.; Teschendorff, A.; Miranda-Saavedra, D.; Rueda, O.M.; Sandoval, J.L.; Vidakovic, A.T.; Saadi, A.; Pharoah, P.; et al. TGFβ induces the formation of tumour-initiating cells in claudinlow breast cancer. Nat. Commun 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; McKnight, S.L. Influence of Metabolism on Epigenetics and Disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Soares, L.M.; He, P.C.; Chun, Y.; Suh, H.; Kim, T.; Buratowski, S. Determinants of histone H3K4 methylation patterns. Mol. Cell 2017, 68, 773–785. [Google Scholar] [CrossRef]
- Loyola, A.; Tagami, H.; Bonaldi, T.; Roche, D.; Quivy, J.P.; Imhof, A.; Nakatani, Y.; Dent, S.Y.; Almouzni, G. The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. 2009, 10, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lorton, B.; Gupta, V.; Shechter, D. A TGF-β-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 2017, 36, 373–386. [Google Scholar] [CrossRef]
- Du, D.; Katsuno, Y.; Meyer, D.; Budi, E.H.; Chen, S.H.; Koeppen, H.; Wang, H.; Akhurst, R.J.; Derynck, R. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial-mesenchymal transition. EMBO Rep. 2018, 19, 135–155. [Google Scholar] [CrossRef] [PubMed]
- De Herreros, A.G.; Peiró, S.; Nassour, M.; Savagner, P.; Herreros, A.G. Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Wu, Z.; Russnes, H.G.; Takagi, S.; Peluffo, G.; Vaske, C.; Zhao, X.; Vollan, H.K.M.; Maruyama, R.; Ekram, M.B.; et al. JARID1B is a luminal lineage-driving oncogene in breast cancer. Cancer Cell 2014, 25, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-J.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 Histone Demethylase Activity by Its Associated Factors. Mol. Cell 2005, 19, 857–864. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.-I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef]
- Ferrari-Amorotti, G.; Fragliasso, V.; Esteki, R.; Prudente, Z.; Soliera, A.R.; Cattelani, S.; Manzotti, G.; Grisendi, G.; Dominici, M.; Pieraccioli, M.; et al. Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion. Cancer Res. 2013, 73, 235–245. [Google Scholar] [CrossRef]
- Rivero, S.; Ceballos-Chávez, M.; Bhattacharya, S.S.; Reyes, J.C.; Reyes, J. HMG20A is required for SNAI1-mediated epithelial to mesenchymal transition. Oncogene 2015, 34, 5264–5276. [Google Scholar] [CrossRef]
- Ramadoss, S.; Chen, X.; Wang, C.-Y. Histone Demethylase KDM6B Promotes Epithelial-Mesenchymal Transition. J. Biol. Chem. 2012, 287, 44508–44517. [Google Scholar] [CrossRef]
- Qi, H.H.; Sarkissian, M.; Hu, G.Q.; Wang, Z.; Bhattacharjee, A.; Gordon, D.B.; Gonzales, M.; Lan, F.; Ongusaha, P.P.; Huarte, M.; et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 2010, 466, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ma, S.; Song, N.; Li, X.; Liu, L.; Yang, S.; Ding, X.; Shan, L.; Zhou, X.; Su, D.; et al. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J. Clin. Investig. 2016, 126, 2205–2220. [Google Scholar] [CrossRef] [PubMed]
- Shao, P.; Liu, Q.; Maina, P.K.; Cui, J.; Bair, T.B.; Li, T.; Umesalma, S.; Zhang, W.; Qi, H.H. Histone demethylase PHF8 promotes epithelial to mesenchymal transition and breast tumorigenesis. Nucleic Acids Res. 2017, 45, 1687–1702. [Google Scholar] [CrossRef]
- Roth, S.Y.; Allis, C. Histone Acetylation and Chromatin Assembly: A Single Escort, Multiple Dances? Cell 1996, 87, 5–8. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA Expression in Invasive Carcinoma of the Breast. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.-I.; Iwase, H. HDAC6 Expression Is Correlated with Better Survival in Breast Cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef]
- Yu, L.; Di, Y.; Xin, L.; Ren, Y.; Liu, X.; Sun, X.; Zhang, W.; Yao, Z.; Yang, J. SND1 acts as a novel gene transcription activator recognizing the conserved Motif domains of Smad promoters, inducing TGFβ1 response and breast cancer metastasis. Oncogene 2017, 36, 3903–3914. [Google Scholar] [CrossRef]
- Liu, L.; Jin, G.; Zhou, X. Modeling the relationship of epigenetic modifications to transcription factor binding. Nucleic Acids Res. 2015, 43, 3873–3885. [Google Scholar] [CrossRef]
- Yu, L.; Liu, X.; Cui, K.; Di, Y.; Xin, L.; Sun, X.; Zhang, W.; Yang, X.; Wei, M.; Yao, Z.; et al. SND1 Acts Downstream of TGFβ1 and Upstream of Smurf1 to Promote Breast Cancer Metastasis. Cancer Res. 2015, 75, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.L.C.; Eichhorn, P.J.A. Deciphering the roles of lncRNAs in breast development and disease. Oncotarget 2018, 9, 20179–20212. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, K.P.; Thomassen, M.; Tan, Q.; Bak, M.; Cold, S.; Burton, M.; Larsen, M.J.; Kruse, T.A. Long non-coding RNA HOTAIR is an independent prognostic marker of metastasis in estrogen receptor-positive primary breast cancer. Breast Cancer Res. Treat. 2013, 142, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Meseure, D.; Vacher, S.; Lallemand, F.; Alsibai, K.D.; Hatem, R.; Chemlali, W.; Nicolas, A.; De Koning, L.; Pasmant, E.; Callens, C.; et al. Prognostic value of a newly identified MALAT1 alternatively spliced transcript in breast cancer. Br. J. Cancer 2016, 114, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef]
- Shi, S.-J.; Wang, L.-J.; Yu, B.; Li, Y.-H.; Jin, Y.; Bai, X.-Z. LncRNA-ATB promotes trastuzumab resistance and invasion-metastasis cascade in breast cancer. Oncotarget 2015, 6, 11652–11663. [Google Scholar] [CrossRef]
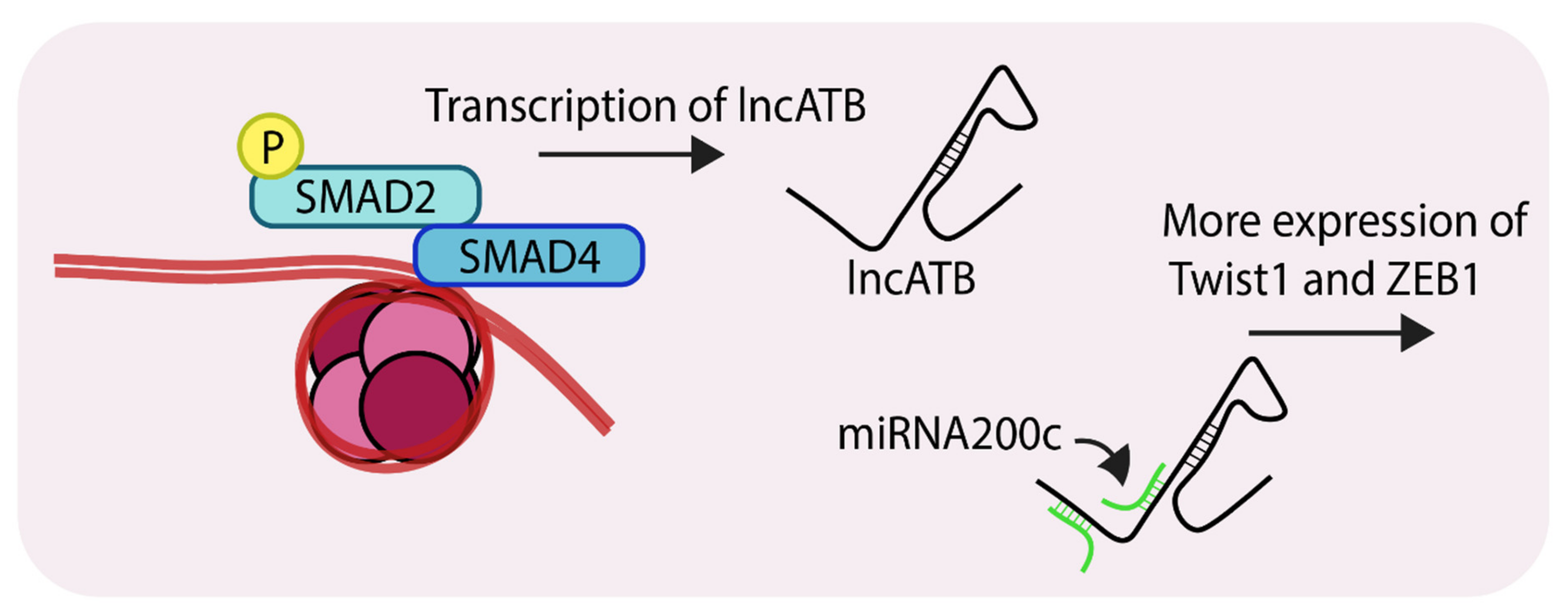
- Li, R.-H.; Chen, M.; Liu, J.; Shao, C.-C.; Guo, C.-P.; Wei, X.-L.; Li, Y.-C.; Huang, W.-H.; Zhang, G.-J. Long noncoding RNA ATB promotes the epithelial–mesenchymal transition by upregulating the miR-200c/Twist1 axe and predicts poor prognosis in breast cancer. Cell Death 2018, 9. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Martin, T.A.; Goyal, A.; Watkins, G.; Jiang, W.G. Expression of the Transcription Factors Snail, Slug, and Twist and Their Clinical Significance in Human Breast Cancer. Ann. Surg. Oncol. 2005, 12, 488–496. [Google Scholar] [CrossRef]
- Guaita, S.; Puig, I.; Garrido, M.; Dominguez, D.; Batlle, E.; Sancho, E.; Dedhar, S.; Baulida, J.; Franci, C.; De Herreros, A.G. Snail Induction of Epithelial to Mesenchymal Transition in Tumor Cells Is Accompanied by MUC1 Repression andZEB1 Expression. J. Biol. Chem. 2002, 277, 39209–39216. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Park, S.I.; McCauley, L.K.; Wang, C.Y. Signaling between transforming growth factor β (TGF-β) and transcription factor SNAI2 represses expression of microRNA miR-203 to promote epithelial-mesenchymal transition and tumor metastasis. J. Biol. Chem. 2013, 288, 10241–10253. [Google Scholar] [CrossRef]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.A.; Heier, A. Induction of Heart Valve Lesions by Small-Molecule ALK5 Inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Fermento, M.E.; Gandini, N.A.; Salomón, D.G.; Ferronato, M.J.; Vitale, C.A.; Arévalo, J.; Romero, A.L.; Nuñez, M.; Jung, M.; Facchinetti, M.M.; et al. Inhibition of p300 suppresses growth of breast cancer. Role of p300 subcellular localization. Exp. Mol. Pathol. 2014, 97, 411–424. [Google Scholar] [CrossRef]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; DiGiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nat. Cell Biol. 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Arun, G.; Diermeier, S.D.; Spector, D.L. Therapeutic Targeting of Long Non-Coding RNAs in Cancer. Trends Mol. Med. 2018, 24, 257–277. [Google Scholar] [CrossRef]
- Panneerdoss, S.; Eedunuri, V.K.; Yadav, P.; Timilsina, S.; Rajamanickam, S.; Viswanadhapalli, S.; Abdelfattah, N.; Onyeagucha, B.C.; Cui, X.; Lai, Z.; et al. Cross-talk among writers, readers, and erasers of m6A regulates cancer growth and progression. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; de Los Mozos, I.R.; Sadée, C.; et al. The SMAD2/3 interactome reveals that TGF-β controls m(6)A mRNA methylation in pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suriyamurthy, S.; Baker, D.; ten Dijke, P.; Iyengar, P.V. Epigenetic Reprogramming of TGF-β Signaling in Breast Cancer. Cancers 2019, 11, 726. https://doi.org/10.3390/cancers11050726
Suriyamurthy S, Baker D, ten Dijke P, Iyengar PV. Epigenetic Reprogramming of TGF-β Signaling in Breast Cancer. Cancers. 2019; 11(5):726. https://doi.org/10.3390/cancers11050726
Chicago/Turabian StyleSuriyamurthy, Sudha, David Baker, Peter ten Dijke, and Prasanna Vasudevan Iyengar. 2019. "Epigenetic Reprogramming of TGF-β Signaling in Breast Cancer" Cancers 11, no. 5: 726. https://doi.org/10.3390/cancers11050726
APA StyleSuriyamurthy, S., Baker, D., ten Dijke, P., & Iyengar, P. V. (2019). Epigenetic Reprogramming of TGF-β Signaling in Breast Cancer. Cancers, 11(5), 726. https://doi.org/10.3390/cancers11050726