Macro Histone Variants: Emerging Rheostats of Gastrointestinal Cancers


{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Gastrointestinal Cancer Epidemiology
1.2. Gastrointestinal Cancer and Epigenetics
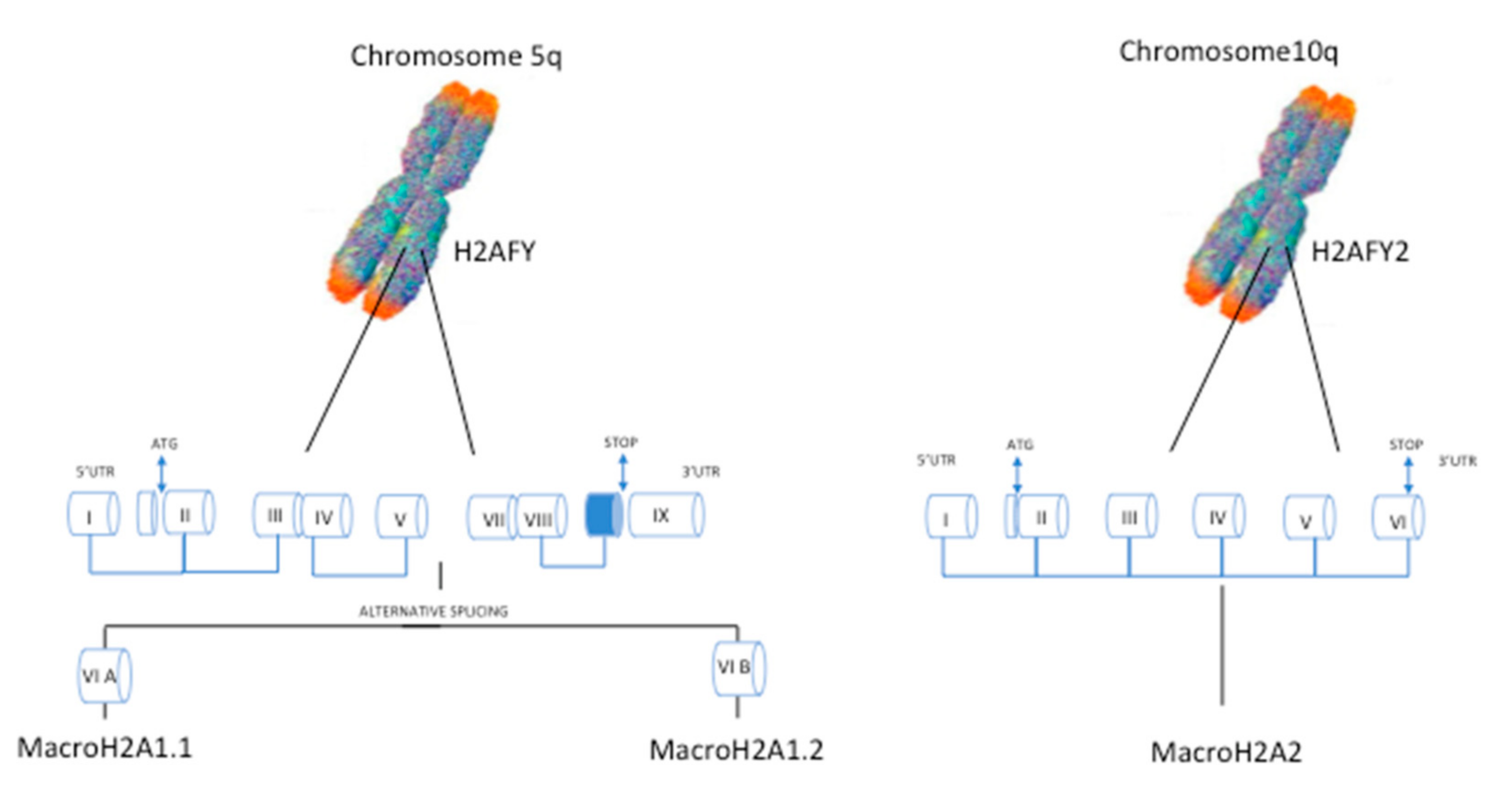
2. The MacroH2A Histone Family Variants
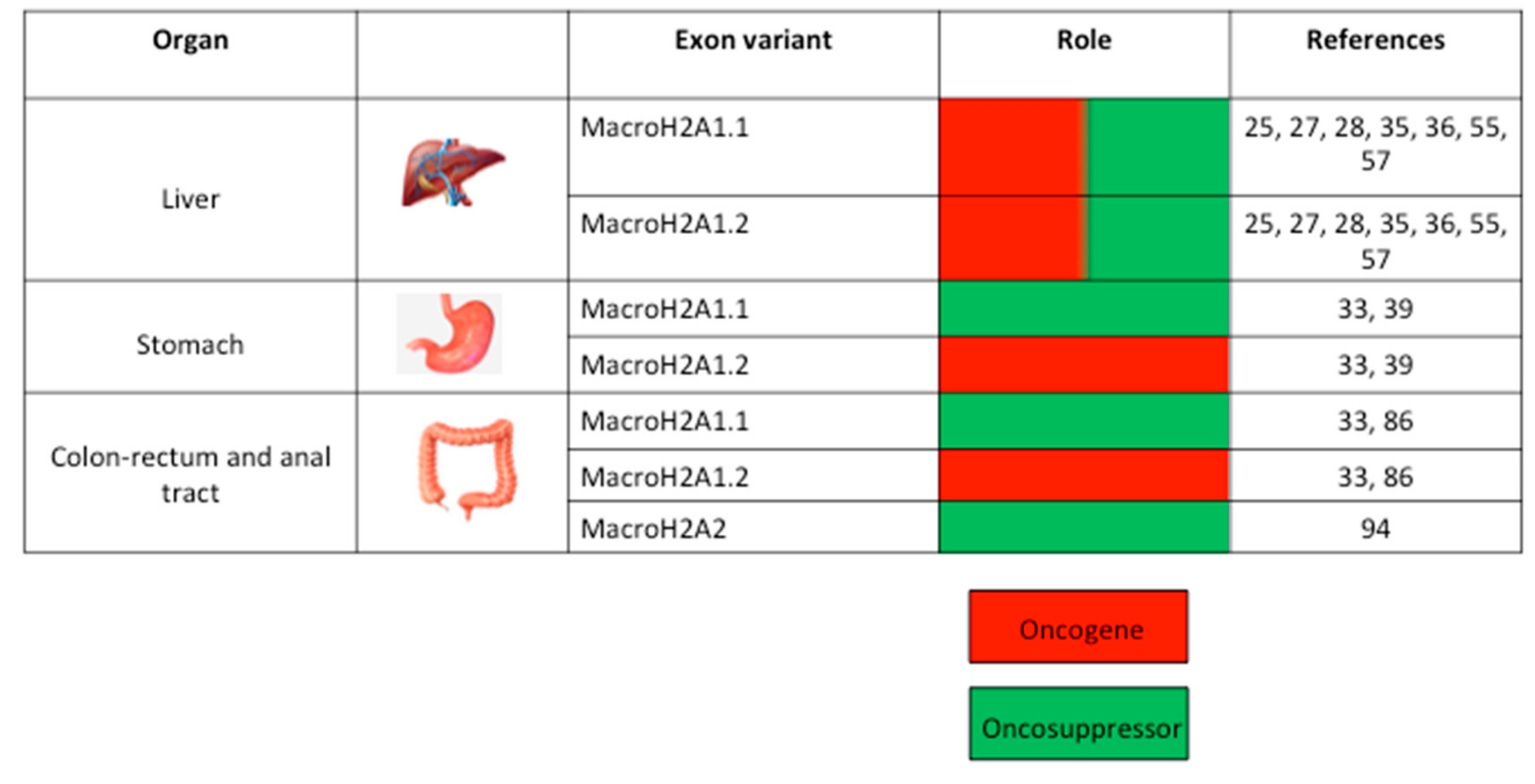
3. MacroH2A and GC of the Upper Digestive Tract
4. MacroH2A and GC of the Hepatobiliary Tree
4.1. MacroH2A1 and NAFLD
4.2. MacroH2A1 and Adipogenesis
4.3. MacroH2A1, Methylation Status, and HCC
5. MacroH2A and GC of the Lower Digestive Tract
5.1. MacroH2A1 and CRC Pathogenesis
5.2. MacroH2A1 Splicing in CRC
5.3. MacroH2A2 in Anal SCC
6. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, L.; Fujimoto, J.; Qiu, D.; Sakamoto, N. Trends in cancer mortality in the elderly in Japan, 1970–2007. Ann. Oncol. 2010, 21, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Bedine, M.S. Textbook of gastroenterology. Gastroenterology 2000, 118, 984–985. [Google Scholar] [CrossRef]
- Pearson-Stuttard, J.; Zhou, B.; Kontis, V.; Bentham, J.; Gunter, M.J.; Ezzati, M. Worldwide burden of cancer attributable to diabetes and high body-mass index: A comparative risk assessment. Lancet Diabetes Endocrinol. 2018, 6, e6–e15. [Google Scholar] [CrossRef]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Jenab, M.; Gunter, M.J. Adiposity and gastrointestinal cancers: Epidemiology, mechanisms and future directions. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 659–670. [Google Scholar] [CrossRef]
- Ulrich, C.M.; Himbert, C.; Holowatyj, A.N.; Hursting, S.D. Energy balance and gastrointestinal cancer: Risk, interventions, outcomes and mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 683–698. [Google Scholar] [CrossRef]
- Forgacs, I.; Ashton, R.; Allum, W.; Bowley, T.; Brown, H.; Coleman, M.P.; Fitzgerald, R.; Glynn, M.; Hiom, S.; Jones, R.; et al. Conference report: Improving outcomes for gastrointestinal cancer in the UK. Front. Gastroenterol. 2018, 9, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Vedeld, H.M.; Goel, A.; Lind, G.E. Epigenetic biomarkers in gastrointestinal cancers: The current state and clinical perspectives. Semin. Cancer Biol. 2018, 51, 36–49. [Google Scholar] [CrossRef]
- Palmirotta, R.; Lovero, D.; Cafforio, P.; Felici, C.; Mannavola, F.; Pelle, E.; Quaresmini, D.; Tucci, M.; Silvestris, F. Liquid biopsy of cancer: A multimodal diagnostic tool in clinical oncology. Ther. Adv. Med. Oncol. 2018, 10, 1758835918794630. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef]
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.O.; Kornberg, R.D. An octamer of histones in chromatin and free in solution. Proce Natl. Acad. Sci. USA 1975, 72, 2626–2630. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Henikoff, S. Nucleosome dynamics during chromatin remodeling in vivo. Nucleus 2016, 7, 20–26. [Google Scholar] [CrossRef][Green Version]
- Talbert, P.B.; Henikoff, S. Histone variants—Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Environmental responses mediated by histone variants. Trends Cell Biol. 2014, 24, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, S. Histone variants in pluripotency and disease. Development 2013, 140, 2513–2524. [Google Scholar] [CrossRef] [PubMed]
- Filipescu, D.; Muller, S.; Almouzni, G. Histone H3 variants and their chaperones during development and disease: Contributing to epigenetic control. Ann. Rev. Cell Dev. Biol. 2014, 30, 615–646. [Google Scholar] [CrossRef] [PubMed]
- Gurard-Levin, Z.A.; Quivy, J.P.; Almouzni, G. Histone chaperones: Assisting histone traffic and nucleosome dynamics. Ann. Rev. Biochem. 2014, 83, 487–517. [Google Scholar] [CrossRef]
- Costanzi, C.; Pehrson, J.R. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 1998, 393, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Casas, C.; Gonzalez-Romero, R.; Cheema, M.S.; Ausio, J.; Eirin-Lopez, J.M. The characterization of macroH2A beyond vertebrates supports an ancestral origin and conserved role for histone variants in chromatin. Epigenetics 2016, 11, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, S.; Gundimella, S.K.; Caron, C.; Perche, P.Y.; Pehrson, J.R.; Khochbin, S.; Luger, K. Structural characterization of the histone variant macroH2A. Mol. Cell. Biol. 2005, 25, 7616–7624. [Google Scholar] [CrossRef] [PubMed]
- Cantarino, N.; Douet, J.; Buschbeck, M. MacroH2A—An epigenetic regulator of cancer. Cancer Lett. 2013, 336, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, O.; Vinciguerra, M. Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration. Genes 2017, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.P.; Huang, T.; Mastrangelo, M.A.; Loring, J.; Panning, B.; Jaenisch, R. Messenger RNAs encoding mouse histone macroH2A1 isoforms are expressed at similar levels in male and female cells and result from alternative splicing. Nucleic Acids Res. 1999, 27, 3685–3689. [Google Scholar] [CrossRef]
- Hernandez-Munoz, I.; Lund, A.H.; van der Stoop, P.; Boutsma, E.; Muijrers, I.; Verhoeven, E.; Nusinow, D.A.; Panning, B.; Marahrens, Y.; van Lohuizen, M. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 7635–7640. [Google Scholar] [CrossRef]
- Nusinow, D.A.; Hernandez-Munoz, I.; Fazzio, T.G.; Shah, G.M.; Kraus, W.L.; Panning, B. Poly (ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, MacroH2A, and contributes to silencing of the inactive X chromosome. J. Biol. Chem. 2007, 282, 12851–12859. [Google Scholar] [CrossRef] [PubMed]
- Mietton, F.; Sengupta, A.K.; Molla, A.; Picchi, G.; Barral, S.; Heliot, L.; Grange, T.; Wutz, A.; Dimitrov, S. Weak but uniform enrichment of the histone variant macroH2A1 along the inactive X chromosome. Mol. Cell. Biol. 2009, 29, 150–156. [Google Scholar] [CrossRef]
- Pazienza, V.; Panebianco, C.; Rappa, F.; Memoli, D.; Borghesan, M.; Cannito, S.; Oji, A.; Mazza, G.; Tamburrino, D.; Fusai, G.; et al. Histone macroH2A1.2 promotes metabolic health and leanness by inhibiting adipogenesis. Epigenet. Chromatin 2016, 9, 45. [Google Scholar] [CrossRef]
- Soma, A.; Sato, K.; Nakanishi, T. Visualization of inactive X chromosome in preimplantation embryos utilizing MacroH2A-EGFP transgenic mouse. Genesis 2013, 51, 259–267. [Google Scholar] [CrossRef]
- Borghesan, M.; Fusilli, C.; Rappa, F.; Panebianco, C.; Rizzo, G.; Oben, J.A.; Mazzoccoli, G.; Faulkes, C.; Pata, I.; Agodi, A.; et al. DNA Hypomethylation and Histone Variant macroH2A1 Synergistically Attenuate Chemotherapy-Induced Senescence to Promote Hepatocellular Carcinoma Progression. Cancer Res. 2016, 76, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, O.; Fusilli, C.; Rappa, F.; Van Haele, M.; Douet, J.; Pindjakova, J.; Rocha, S.W.; Pata, I.; Valcikova, B.; Uldrijan, S.; et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology 2017. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; Leroy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Creppe, C.; Janich, P.; Cantarino, N.; Noguera, M.; Valero, V.; Musulen, E.; Douet, J.; Posavec, M.; Martin-Caballero, J.; Sumoy, L.; et al. MacroH2A1 regulates the balance between self-renewal and differentiation commitment in embryonic and adult stem cells. Mol. Cell. Biol. 2012, 32, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Pierredon, S.; Driouch, K.; Gratadou, L.; Lacroix-Triki, M.; Espinoza, M.P.; Zonta, E.; Germann, S.; Mortada, H.; Villemin, J.P.; et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 2012, 19, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Shim, J.W.; Park, H.S.; Eum, D.Y.; Park, M.T.; Mi Yi, J.; Choi, S.H.; Kim, S.D.; Son, T.G.; Lu, W.; et al. MacroH2A1 downregulation enhances the stem-like properties of bladder cancer cells by transactivation of Lin28B. Oncogene 2016, 35, 1292–1301. [Google Scholar] [CrossRef]
- Novikov, L.; Park, J.W.; Chen, H.; Klerman, H.; Jalloh, A.S.; Gamble, M.J. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol. Cell. Biol. 2011, 31, 4244–4255. [Google Scholar] [CrossRef]
- Sporn, J.C.; Kustatscher, G.; Hothorn, T.; Collado, M.; Serrano, M.; Muley, T.; Schnabel, P.; Ladurner, A.G. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 2009, 28, 3423–3428. [Google Scholar] [CrossRef] [PubMed]
- Jueliger, S.; Lyons, J.; Cannito, S.; Pata, I.; Pata, P.; Shkolnaya, M.; Lo Re, O.; Peyrou, M.; Villarroya, F.; Pazienza, V.; et al. Efficacy and epigenetic interactions of novel DNA hypomethylating agent guadecitabine (SGI-110) in preclinical models of hepatocellular carcinoma. Epigenetics 2016, 11, 709–720. [Google Scholar] [CrossRef]
- Rappa, F.; Greco, A.; Podrini, C.; Cappello, F.; Foti, M.; Bourgoin, L.; Peyrou, M.; Marino, A.; Scibetta, N.; Williams, R.; et al. Immunopositivity for histone macroH2A1 isoforms marks steatosis-associated hepatocellular carcinoma. PloS ONE 2013, 8, e54458. [Google Scholar] [CrossRef]
- Ku, G.Y.; Ilson, D.H. Preface on Esophagus Cancer. Chin. Clin. Oncol. 2017, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Brenner, H.; Rothenbacher, D.; Arndt, V. Epidemiology of stomach cancer. Methods Mol. Biol. 2009, 472, 467–477. [Google Scholar] [CrossRef]
- Li, F.; Yi, P.; Pi, J.; Li, L.; Hui, J.; Wang, F.; Liang, A.; Yu, J. QKI5-mediated alternative splicing of the histone variant macroH2A1 regulates gastric carcinogenesis. Oncotarget 2016, 7, 32821–32834. [Google Scholar] [CrossRef] [PubMed]
- Keplinger, K.M.; Bloomston, M. Anatomy and embryology of the biliary tract. Surg. Clin. North Am. 2014, 94, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.L. Bile formation and secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [CrossRef] [PubMed]
- Benavides, M.; Anton, A.; Gallego, J.; Gomez, M.A.; Jimenez-Gordo, A.; La Casta, A.; Laquente, B.; Macarulla, T.; Rodriguez-Mowbray, J.R.; Maurel, J. Biliary tract cancers: SEOM clinical guidelines. Clin. Transl. Oncol. 2015, 17, 982–987. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Global Burden of Disease Liver Cancer, C.; Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef]
- Sangiovanni, A.; Prati, G.M.; Fasani, P.; Ronchi, G.; Romeo, R.; Manini, M.; Del Ninno, E.; Morabito, A.; Colombo, M. The natural history of compensated cirrhosis due to hepatitis C virus: A 17-year cohort study of 214 patients. Hepatology 2006, 43, 1303–1310. [Google Scholar] [CrossRef]
- Podrini, C.; Borghesan, M.; Greco, A.; Pazienza, V.; Mazzoccoli, G.; Vinciguerra, M. Redox homeostasis and epigenetics in non-alcoholic fatty liver disease (NAFLD). Curr. Pharm. Des. 2013, 19, 2737–2746. [Google Scholar] [CrossRef]
- Changolkar, L.N.; Singh, G.; Cui, K.; Berletch, J.B.; Zhao, K.; Disteche, C.M.; Pehrson, J.R. Genome-wide distribution of macroH2A1 histone variants in mouse liver chromatin. Mol. Cell. Biol. 2010, 30, 5473–5483. [Google Scholar] [CrossRef] [PubMed]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lv, P.; Yan, G.; Fan, H.; Cheng, L.; Zhang, F.; Dang, Y.; Wu, H.; Wen, B. MacroH2A1 associates with nuclear lamina and maintains chromatin architecture in mouse liver cells. Sci. Rep. 2015, 5, 17186. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Invest. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Borghesan, M.; Mazzoccoli, G.; Sheedfar, F.; Oben, J.; Pazienza, V.; Vinciguerra, M. Histone variants and lipid metabolism. Biochem. Soc. Trans. 2014, 42, 1409–1413. [Google Scholar] [CrossRef]
- Sheedfar, F.; Vermeer, M.; Pazienza, V.; Villarroya, J.; Rappa, F.; Cappello, F.; Mazzoccoli, G.; Villarroya, F.; van der Molen, H.; Hofker, M.H.; et al. Genetic ablation of macrohistone H2A1 leads to increased leanness, glucose tolerance and energy expenditure in mice fed a high-fat diet. Int. J. Obes. 2015, 39, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Changolkar, L.N.; Costanzi, C.; Leu, N.A.; Chen, D.; McLaughlin, K.J.; Pehrson, J.R. Developmental changes in histone macroH2A1-mediated gene regulation. Mol. Cell. Biol. 2007, 27, 2758–2764. [Google Scholar] [CrossRef] [PubMed]
- Boulard, M.; Storck, S.; Cong, R.; Pinto, R.; Delage, H.; Bouvet, P. Histone variant macroH2A1 deletion in mice causes female-specific steatosis. Epigenet. Chromatin 2010, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Pazienza, V.; Borghesan, M.; Mazza, T.; Sheedfar, F.; Panebianco, C.; Williams, R.; Mazzoccoli, G.; Andriulli, A.; Nakanishi, T.; Vinciguerra, M. SIRT1-metabolite binding histone macroH2A1.1 protects hepatocytes against lipid accumulation. Aging 2014, 6, 35–47. [Google Scholar] [CrossRef]
- Wan, D.; Liu, C.; Sun, Y.; Wang, W.; Huang, K.; Zheng, L. MacroH2A1.1 cooperates with EZH2 to promote adipogenesis by regulating Wnt signaling. J. Mol. Cell Biol. 2017, 9, 325–337. [Google Scholar] [CrossRef]
- Podrini, C.; Koffas, A.; Chokshi, S.; Vinciguerra, M.; Lelliott, C.J.; White, J.K.; Adissu, H.A.; Williams, R.; Greco, A. MacroH2A1 isoforms are associated with epigenetic markers for activation of lipogenic genes in fat-induced steatosis. FASEB J. 2015, 29, 1676–1687. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Pehrson, J.R.; Changolkar, L.N.; Costanzi, C.; Leu, N.A. Mice without macroH2A histone variants. Mol. Cell. Biol. 2014, 34, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Sheedfar, F.; Di Biase, S.; Koonen, D.; Vinciguerra, M. Liver diseases and aging: Friends or foes? Aging Cell 2013, 12, 950–954. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Lee, J.S.; Conner, E.A.; Schroeder, I.; Factor, V.M.; Thorgeirsson, S.S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Invest. 2007, 117, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Sceusi, E.L.; Loose, D.S.; Wray, C.J. Clinical implications of DNA methylation in hepatocellular carcinoma. HPB 2011, 13, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, O.; Douet, J.; Buschbeck, M.; Fusilli, C.; Pazienza, V.; Panebianco, C.; Castracani, C.C.; Mazza, T.; Li Volti, G.; Vinciguerra, M. Histone variant macroH2A1 rewires carbohydrate and lipid metabolism of hepatocellular carcinoma cells towards cancer stem cells. Epigenetics 2018, 13, 829–845. [Google Scholar] [CrossRef]
- Helander, H.F.; Fandriks, L. Surface area of the digestive tract—Revisited. Scand. J. Gastroenterol. 2014, 49, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Clevers, H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef]
- Yan, K.S.; Chia, L.A.; Li, X.; Ootani, A.; Su, J.; Lee, J.Y.; Su, N.; Luo, Y.; Heilshorn, S.C.; Amieva, M.R.; et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc. Natl. Acad. Sci. USA 2012, 109, 466–471. [Google Scholar] [CrossRef]
- Powell, A.E.; Wang, Y.; Li, Y.; Poulin, E.J.; Means, A.L.; Washington, M.K.; Higginbotham, J.N.; Juchheim, A.; Prasad, N.; Levy, S.E.; et al. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 2012, 149, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Cedeno, R.J.; Nakauka-Ddamba, A.; Yousefi, M.; Sterling, S.; Leu, N.A.; Li, N.; Pehrson, J.R.; Lengner, C.J. The histone variant macroH2A confers functional robustness to the intestinal stem cell compartment. PLoS ONE 2017, 12, e0185196. [Google Scholar] [CrossRef]
- Yu, D.H.; Gadkari, M.; Zhou, Q.; Yu, S.; Gao, N.; Guan, Y.; Schady, D.; Roshan, T.N.; Chen, M.H.; Laritsky, E.; et al. Postnatal epigenetic regulation of intestinal stem cells requires DNA methylation and is guided by the microbiome. Genome Biol. 2015, 16, 211. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G. Colorectal Cancer: Genetic Abnormalities, Tumor Progression, Tumor Heterogeneity, Clonal Evolution and Tumor-Initiating Cells. Med. Sci. 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.W.; DiSario, J.A.; Cannon-Albright, L. Genetics of colon cancer: Impact of inheritance on colon cancer risk. Ann. Rev. Med. 1995, 46, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Smyrk, T.C.; Watson, P.; Lanspa, S.J.; Lynch, J.F.; Lynch, P.M.; Cavalieri, R.J.; Boland, C.R. Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: An updated review. Gastroenterology 1993, 104, 1535–1549. [Google Scholar] [CrossRef]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Barzily-Rokni, M.; Friedman, N.; Ron-Bigger, S.; Isaac, S.; Michlin, D.; Eden, A. Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A). Nucleic Acids Res. 2011, 39, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Vardabasso, C.; Hasson, D.; Ratnakumar, K.; Chung, C.Y.; Duarte, L.F.; Bernstein, E. Histone variants: Emerging players in cancer biology. Cell Mol. Life Sci. 2014, 71, 379–404. [Google Scholar] [CrossRef] [PubMed]
- De Barrios, O.; Gyorffy, B.; Fernandez-Acenero, M.J.; Sanchez-Tillo, E.; Sanchez-Moral, L.; Siles, L.; Esteve-Arenys, A.; Roue, G.; Casal, J.I.; Darling, D.S.; et al. ZEB1-induced tumourigenesis requires senescence inhibition via activation of DKK1/mutant p53/Mdm2/CtBP and repression of macroH2A1. Gut 2017, 66, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell Mol. Life Sci 2012, 69, 3429–3456. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, M.; Ling, H.; Ivan, C.; Pichler, M.; Matsushita, D.; Goblirsch, M.; Stiegelbauer, V.; Shigeyasu, K.; Zhang, X.; Chen, M.; et al. H19 Noncoding RNA, an Independent Prognostic Factor, Regulates Essential Rb-E2F and CDK8-beta-Catenin Signaling in Colorectal Cancer. EBioMedicine 2016, 13, 113–124. [Google Scholar] [CrossRef]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423. [Google Scholar] [CrossRef]
- Castle, J.C.; Zhang, C.; Shah, J.K.; Kulkarni, A.V.; Kalsotra, A.; Cooper, T.A.; Johnson, J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008, 40, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Fu, H.; Zhang, J.; Lu, X.; Yu, F.; Jin, L.; Bai, L.; Huang, B.; Shen, L.; Feng, Y.; et al. RNA-binding protein quaking, a critical regulator of colon epithelial differentiation and a suppressor of colon cancer. Gastroenterology 2010, 138, 231–240. [Google Scholar] [CrossRef]
- Yin, D.; Ogawa, S.; Kawamata, N.; Tunici, P.; Finocchiaro, G.; Eoli, M.; Ruckert, C.; Huynh, T.; Liu, G.; Kato, M.; et al. High-resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol. Cancer Res. 2009, 7, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef]
- Sporn, J.C.; Jung, B. Differential regulation and predictive potential of MacroH2A1 isoforms in colon cancer. Am. J. Pathol. 2012, 180, 2516–2526. [Google Scholar] [CrossRef] [PubMed]
- Warth, A.; Cortis, J.; Soltermann, A.; Meister, M.; Budczies, J.; Stenzinger, A.; Goeppert, B.; Thomas, M.; Herth, F.J.; Schirmacher, P.; et al. Tumour cell proliferation (Ki-67) in non-small cell lung cancer: A critical reappraisal of its prognostic role. Br. J. Cancer 2014, 111, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar]
- Islami, F.; Ferlay, J.; Lortet-Tieulent, J.; Bray, F.; Jemal, A. International trends in anal cancer incidence rates. Int. J. Epidemiol. 2017, 46, 924–938. [Google Scholar] [CrossRef] [PubMed]
- P.H.A.o. Canada. Section 5–5: Canadian Guidelines on Sexually Transmitted Infections—Management and Treatment of Specific Infections—Human Papillomavirus (HPV) infections—Canada.ca; Government of Canada: Ontario, ON, Canada, 2018.
- Hoedema, R.E. Anal Intraepithelial Neoplasia and Squamous Cell Cancer of the Anus. Clin. Colon. Rectal. Surg. 2018, 31, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Uronis, H.E.; Bendell, J.C. Anal cancer: An overview. Oncologist 2007, 12, 524–534. [Google Scholar] [CrossRef]
- Hu, W.H.; Miyai, K.; Sporn, J.C.; Luo, L.; Wang, J.Y.; Cosman, B.; Ramamoorthy, S. Loss of histone variant macroH2A2 expression associates with progression of anal neoplasm. J. Clin. Pathol. 2016, 69, 627–631. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giallongo, S.; Lo Re, O.; Vinciguerra, M. Macro Histone Variants: Emerging Rheostats of Gastrointestinal Cancers. Cancers 2019, 11, 676. https://doi.org/10.3390/cancers11050676
Giallongo S, Lo Re O, Vinciguerra M. Macro Histone Variants: Emerging Rheostats of Gastrointestinal Cancers. Cancers. 2019; 11(5):676. https://doi.org/10.3390/cancers11050676
Chicago/Turabian StyleGiallongo, Sebastiano, Oriana Lo Re, and Manlio Vinciguerra. 2019. "Macro Histone Variants: Emerging Rheostats of Gastrointestinal Cancers" Cancers 11, no. 5: 676. https://doi.org/10.3390/cancers11050676
APA StyleGiallongo, S., Lo Re, O., & Vinciguerra, M. (2019). Macro Histone Variants: Emerging Rheostats of Gastrointestinal Cancers. Cancers, 11(5), 676. https://doi.org/10.3390/cancers11050676