Unleashing Tumour-Dendritic Cells to Fight Cancer by Tackling Their Three A’s: Abundance, Activation and Antigen-Delivery

 ,
,
Abstract
1. Introduction
2. DC Subsets: Adding Another Layer of Complexity to DC-Based Therapies
3. Abundance: Increasing DC Infiltration into Tumours
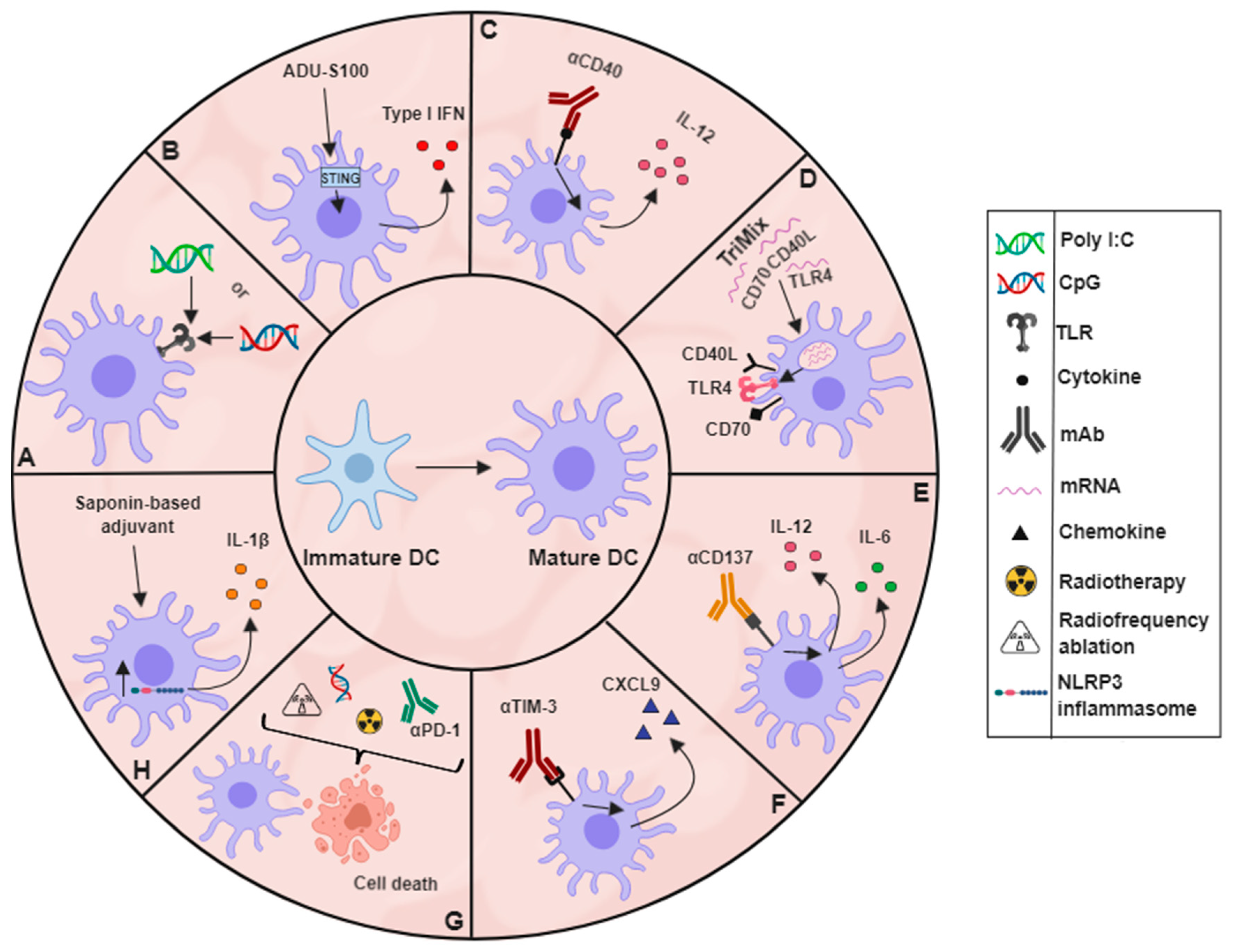
4. Activation: Maturing DCs Residing within Tumours
5. Antigen Delivery: Steering the Immune Response
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- Schuler, G.; Schuler-Thurner, B.; Steinman, R.M. The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 2003, 15, 138–147. [Google Scholar] [CrossRef]
- O’Keeffe, M.; Mok, W.H.; Radford, K.J. Human dendritic cell subsets and function in health and disease. Cell. Mol. Life Sci. 2015, 72, 4309–4325. [Google Scholar] [CrossRef]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, C.-A.; Wang, L.-F.; Ginhoux, F. Aligning bona fide dendritic cell populations across species. Cell. Immunol. 2014, 291, 3–10. [Google Scholar] [CrossRef]
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat. Commun. 2016, 7, 13720. [Google Scholar] [CrossRef]
- Onai, N.; Kurabayashi, K.; Hosoi-Amaike, M.; Toyama-Sorimachi, N.; Matsushima, K.; Inaba, K.; Ohteki, T. A Clonogenic Progenitor with Prominent Plasmacytoid Dendritic Cell Developmental Potential. Immunity 2013, 38, 943–957. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Hochrein, H.; O’Keeffe, M.; Wagner, H. Human and mouse plasmacytoid dendritic cells. Hum. Immunol. 2002, 63, 1103–1110. [Google Scholar] [CrossRef]
- Schlitzer, A.; Sivakamasundari, V.; Chen, J.; Sumatoh, H.R.B.; Schreuder, J.; Lum, J.; Malleret, B.; Zhang, S.; Larbi, A.; Zolezzi, F.; et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat. Immunol. 2015, 16, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Dutertre, C.-A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Bakdash, G.; Buschow, S.I.; Gorris, M.A.J.; Halilovic, A.; Hato, S.V.; Sköld, A.E.; Schreibelt, G.; Sittig, S.P.; Torensma, R.; Duiveman-de Boer, T.; et al. Expansion of a BDCA1+CD14+ Myeloid Cell Population in Melanoma Patients May Attenuate the Efficacy of Dendritic Cell Vaccines. Cancer Res. 2016, 76, 4332–4346. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.M.; Liu, Y.-J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Valladeau-Guilemond, J.; Donnadieu, M.-H.; Sastre-Garau, X.; Soumelis, V.; Amigorena, S. Characterization of resident and migratory dendritic cells in human lymph nodes. J. Exp. Med. 2012, 209, 653–660. [Google Scholar] [CrossRef]
- Schlitzer, A.; Ginhoux, F. Organization of the mouse and human DC network. Curr. Opin. Immunol. 2014, 26, 90–99. [Google Scholar] [CrossRef]
- Pulendran, B.; Banchereau, J.; Burkeholder, S.; Kraus, E.; Guinet, E.; Chalouni, C.; Caron, D.; Maliszewski, C.; Davoust, J.; Fay, J.; et al. Flt3-ligand and granulocyte colony-stimulating factor mobilize distinct human dendritic cell subsets in vivo. J. Immunol. 2000, 165, 566–572. [Google Scholar] [CrossRef]
- Haniffa, M.; Shin, A.; Bigley, V.; McGovern, N.; Teo, P.; See, P.; Wasan, P.S.; Wang, X.-N.; Malinarich, F.; Malleret, B.; et al. Human Tissues Contain CD141hi Cross-Presenting Dendritic Cells with Functional Homology to Mouse CD103+ Nonlymphoid Dendritic Cells. Immunity 2012, 37, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Jaensson, E.; Uronen-Hansson, H.; Pabst, O.; Eksteen, B.; Tian, J.; Coombes, J.L.; Berg, P.-L.; Davidsson, T.; Powrie, F.; Johansson-Lindbom, B.; et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J. Exp. Med. 2008, 205, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Haniffa, M.; Collin, M.; Ginhoux, F. Ontogeny and Functional Specialization of Dendritic Cells in Human and Mouse. Adv. Immunol. 2013, 120, 1–49. [Google Scholar]
- Schlitzer, A.; McGovern, N.; Teo, P.; Zelante, T.; Atarashi, K.; Low, D.; Ho, A.W.S.; See, P.; Shin, A.; Wasan, P.S.; et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 2013, 38, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, S.; Salem, S.; Bustamante, J.; Bigley, V.; Boisson-Dupuis, S.; Azevedo, J.; Fortin, A.; Haniffa, M.; Ceron-Gutierrez, L.; Bacon, C.; et al. Mutations in IRF8 and Human Dendritic Cell Immunodeficiency. N. Engl. J. Med. 2011, 365, 127. [Google Scholar] [CrossRef]
- Mittag, D.; Proietto, A.I.; Loudovaris, T.; Mannering, S.I.; Vremec, D.; Shortman, K.; Wu, L.; Harrison, L.C. Human Dendritic Cell Subsets from Spleen and Blood Are Similar in Phenotype and Function but Modified by Donor Health Status. J. Immunol. 2011, 186, 6207–6217. [Google Scholar] [CrossRef]
- Segura, E.; Durand, M.; Amigorena, S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J. Exp. Med. 2013, 210, 1035–1047. [Google Scholar] [CrossRef]
- Nizzoli, G.; Krietsch, J.; Weick, A.; Steinfelder, S.; Facciotti, F.; Gruarin, P.; Bianco, A.; Steckel, B.; Moro, M.; Crosti, M.; et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood 2013, 122, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Galon, J.; Pages, F.; Tartour, E.; Sautes-Fridman, C.; Kroemer, G. Prognostic and Predictive Impact of Intra- and Peritumoral Immune Infiltrates. Cancer Res. 2011, 71, 5601–5605. [Google Scholar] [CrossRef] [PubMed]
- Vicari, A.P.; Caux, C.; Trinchieri, G. Tumour escape from immune surveillance through dendritic cell inactivation. Semin. Cancer Biol. 2002, 12, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Goc, J.; Germain, C.; Vo-Bourgais, T.K.D.; Lupo, A.; Klein, C.; Knockaert, S.; de Chaisemartin, L.; Ouakrim, H.; Becht, E.; Alifano, M.; et al. Dendritic Cells in Tumor-Associated Tertiary Lymphoid Structures Signal a Th1 Cytotoxic Immune Contexture and License the Positive Prognostic Value of Infiltrating CD8+ T Cells. Cancer Res. 2014, 74, 705–715. [Google Scholar] [CrossRef]
- Hargadon, K.M. Tumor-altered dendritic cell function: Implications for anti-tumor immunity. Front. Immunol. 2013, 4, 192. [Google Scholar] [CrossRef] [PubMed]
- Chevolet, I.; Speeckaert, R.; Schreuer, M.; Neyns, B.; Krysko, O.; Bachert, C.; Van Gele, M.; van Geel, N.; Brochez, L. Clinical significance of plasmacytoid dendritic cells and myeloid-derived suppressor cells in melanoma. J. Transl. Med. 2015, 13, 9. [Google Scholar] [CrossRef]
- Tucci, M.; Stucci, L.S.; Mannavola, F.; Passarelli, A.; D’Oronzo, S.; Lospalluti, L.; Giudice, G.; Silvestris, F. Defective levels of both circulating dendritic cells and T-regulatory cells correlate with risk of recurrence in cutaneous melanoma. Clin. Transl. Oncol. 2018, 1–10. [Google Scholar] [CrossRef]
- Failli, A.; Legitimo, A.; Orsini, G.; Romanini, A.; Consolini, R. Numerical defect of circulating dendritic cell subsets and defective dendritic cell generation from monocytes of patients with advanced melanoma. Cancer Lett. 2013, 337, 184–192. [Google Scholar] [CrossRef]
- Lissoni, P.; Vigore, L.; Ferranti, R.; Bukovec, R.; Meregalli, S.; Mandala, M.; Barni, S.; Tancini, G.; Fumagalli, L.; Giani, L. Circulating dendritic cells in early and advanced cancer patients: Diminished percent in the metastatic disease. J. Biol. Regul. Homeost. Agents 1999, 13, 216–219. [Google Scholar]
- Legitimo, A.; Consolini, R.; Failli, A.; Orsini, G.; Spisni, R. Dendritic cell defects in the colorectal cancer. Hum. Vaccin. Immunother. 2014, 10, 3224–3235. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, U.K.; Rutkowski, M.R.; Rauwerdink, A.M.; Fields, J.; Escovar-Fadul, X.; Baird, J.; Cubillos-Ruiz, J.R.; Jacobs, A.C.; Gonzalez, J.L.; Weaver, J.; et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 2012, 209, 495–506. [Google Scholar] [CrossRef]
- Truxova, I.; Kasikova, L.; Hensler, M.; Skapa, P.; Laco, J.; Pecen, L.; Belicova, L.; Praznovec, I.; Halaska, M.J.; Brtnicky, T.; et al. Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J. Immunother. Cancer 2018, 6, 139. [Google Scholar] [CrossRef]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161.e7. [Google Scholar] [CrossRef]
- Waskow, C.; Liu, K.; Darrasse-Jèze, G.; Guermonprez, P.; Ginhoux, F.; Merad, M.; Shengelia, T.; Yao, K.; Nussenzweig, M. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat. Immunol. 2008, 9, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Van de Laar, L.; Coffer, P.J.; Woltman, A.M. Regulation of dendritic cell development by GM-CSF: Molecular control and implications for immune homeostasis and therapy. Blood 2012, 119, 3383–3393. [Google Scholar] [CrossRef]
- Nemunaitis, J. Vaccines in cancer: GVAX®, a GM-CSF gene vaccine. Expert Rev. Vaccines 2005, 4, 259–274. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef]
- Le, D.T.; Ko, A.H.; Wainberg, Z.A.; Picozzi, V.J.; Kindler, H.L.; Wang-Gillam, A.; Oberstein, P.E.; Morse, M.; Zeh, H.; Weekes, C.D.; et al. Results from a phase 2b, randomized, multicenter study of GVAX pancreas and CRS-207 compared to chemotherapy in adults with previously-treated metastatic pancreatic adenocarcinoma (ECLIPSE Study). J. Clin. Oncol. 2017, 35, 345. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1028.e14. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Paulete, A.R.; Teijeira, A.; Quetglas, J.I.; Rodriguez-Ruiz, M.E.; Sanchez-Arraez, A.; Labiano, S.; Etxeberria, I.; Azpilikueta, A.; Bolaños, E.; Ballesteros-Briones, M.C.; et al. Intratumoral immunotherapy with XCL1 and sFlt3L encoded in recombinant Semliki Forest Virus-derived vectors fosters dendritic cell-mediated T cell cross-priming. Cancer Res. 2018, 78, 6643–6654. [Google Scholar] [CrossRef]
- Cook, S.J.; Lee, Q.; Wong, A.C.H.; Spann, B.C.; Vincent, J.N.; Wong, J.J.L.; Schlitzer, A.; Gorrell, M.D.; Weninger, W.; Roediger, B. Differential chemokine receptor expression and usage by pre-cDC1 and pre-cDC2. Immunol. Cell Biol. 2018, 96, 1131–1139. [Google Scholar] [CrossRef]
- Meyer, M.A.; Baer, J.M.; Knolhoff, B.L.; Nywening, T.M.; Panni, R.Z.; Su, X.; Weilbaecher, K.N.; Hawkins, W.G.; Ma, C.; Fields, R.C.; et al. Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat. Commun. 2018, 9, 1250. [Google Scholar] [CrossRef]
- Aznar, M.A.; Tinari, N.; Rullán, A.J.; Sánchez-Paulete, A.R.; Rodriguez-Ruiz, M.E.; Melero, I. Intratumoral Delivery of Immunotherapy—Act Locally, Think Globally. J. Immunol. 2017, 198, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Gu, H.; Tang, M.; Zhao, J.; Cattral, M.S. Tumor Dendritic Cells (DCs) Derived from Precursors of Conventional DCs Are Dispensable for Intratumor CTL Responses. J. Immunol. 2018, 201, 1306–1314. [Google Scholar] [CrossRef]
- Krzastek, S.C.; Goliadze, E.; Zhou, S.; Petrossian, A.; Youniss, F.; Sundaresan, G.; Wang, L.; Zweit, J.; Guruli, G. Dendritic cell trafficking in tumor-bearing mice. Cancer Immunol. Immunother. 2018, 67, 1939–1947. [Google Scholar] [CrossRef]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef]
- Dudek, A.M.; Martin, S.; Garg, A.D.; Agostinis, P. Immature, Semi-Mature, and Fully Mature Dendritic Cells: Toward a DC-Cancer Cells Interface That Augments Anticancer Immunity. Front. Immunol. 2013, 4, 438. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Qu, S.; Chen, X.; Wu, Q.; Shi, M. Promising Targets for Cancer Immunotherapy: TLRs, RLRs, and STING-Mediated Innate Immune Pathways. Int. J. Mol. Sci. 2017, 18, 404. [Google Scholar] [CrossRef]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Adams, S. Toll-like receptor agonists in cancer therapy. Immunotherapy 2009, 1, 949–964. [Google Scholar] [CrossRef]
- Ammi, R.; De Waele, J.; Willemen, Y.; Van Brussel, I.; Schrijvers, D.M.; Lion, E.; Smits, E.L.J. Poly(I:C) as cancer vaccine adjuvant: Knocking on the door of medical breakthroughs. Pharmacol. Ther. 2015, 146, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Clappaert, E.J.; Murgaski, A.; Van Damme, H.; Kiss, M.; Laoui, D. Diamonds in the Rough: Harnessing Tumor-Associated Myeloid Cells for Cancer Therapy. Front. Immunol. 2018, 9, 2250. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Metellus, P.; Ursu, R.; Zohar, S.; Lafitte, F.; Barrie, M.; Meng, Y.; Richard, M.; Parizot, C.; Laigle-Donadey, F.; et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: A phase II study. Neuro-Oncology 2010, 12, 401–408. [Google Scholar] [CrossRef]
- Humbert, M.; Guery, L.; Brighouse, D.; Lemeille, S.; Hugues, S. Intratumoral CpG-B promotes antitumoral neutrophil, CDC, and T-cell cooperation without reprograming tolerogenic pDC. Cancer Res. 2018, 78, 3280–3292. [Google Scholar]
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ohkuri, T.; Ghosh, A.; Kosaka, A.; Zhu, J.; Ikeura, M.; David, M.; Watkins, S.C.; Sarkar, S.N.; Okada, H. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 1199–1208. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Foote, J.B.; Kok, M.; Leatherman, J.M.; Armstrong, T.D.; Marcinkowski, B.C.; Ojalvo, L.S.; Kanne, D.B.; Jaffee, E.M.; Dubensky, T.W., Jr.; Emens, L.A. A STING agonist given with OX40 receptor and PD-L1 modulators primes immunity and reduces tumor growth in tolerized mice. Cancer Immunol. Res. 2018, 5, 468–479. [Google Scholar]
- Ma, D.Y.; Clark, E.A. The role of CD40 and CD154/CD40L in dendritic cells. Semin. Immunol. 2009, 21, 265–272. [Google Scholar] [CrossRef]
- Cella, M.; Scheidegger, D.; Palmer-Lehmann, K.; Lane, P.; Lanzavecchia, A.; Alber, G. Ligation of CD40 on Dendritic Cells Triggers Production of High Levels of Interleukin-12 and Enhances T Cell Stimulatory Capacity: T-T Help via APC Activation. J. Exp. Med. 1996, 184, 747–752. [Google Scholar] [CrossRef]
- Bajor, D.L.; Mick, R.; Riese, M.J.; Huang, A.C.; Sullivan, B.; Richman, L.P.; Torigian, D.A.; George, S.M.; Stelekati, E.; Chen, F.; et al. Long-term outcomes of a phase I study of agonist CD40 antibody and CTLA-4 blockade in patients with metastatic melanoma. Oncoimmunology 2018, 7, e1468956. [Google Scholar] [CrossRef]
- Rech, A.J.; Dada, H.; Kotzin, J.J.; Henao-Mejia, J.; Minn, A.J.; Twyman-Saint Victor, C.; Vonderheide, R.H. Radiotherapy and CD40 Activation Separately Augment Immunity to Checkpoint Blockade in Cancer. Cancer Res. 2018, 78, 4282–4291. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Du Four, S.; Van der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix mRNA Results in T-cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol. Res. 2016, 4, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients With Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Seremet, T.; Koch, A.; Jansen, Y.; Schreuer, M.; Wilgenhof, S.; Del Marmol, V.; Liènard, D.; Thielemans, K.; Schats, K.; Kockx, M.; et al. Molecular and epigenetic features of melanomas and tumor immune microenvironment linked to durable remission to ipilimumab-based immunotherapy in metastatic patients. J. Transl. Med. 2016, 14, 232. [Google Scholar] [CrossRef]
- Kuang, Y.; Chen, H.; Weng, X.; Chen, Z.; Liu, X.; Zhu, H. Effects of 4-1BB signaling on the biological function of murine dendritic cells. Oncol. Lett. 2011, 3, 477–481. [Google Scholar] [CrossRef]
- Murillo, O.; Dubrot, J.; Palazón, A.; Arina, A.; Azpilikueta, A.; Alfaro, C.; Solano, S.; Ochoa, M.C.; Berasain, C.; Gabari, I.; et al. In vivo depletion of DC impairs the anti-tumor effect of agonistic anti-CD137 mAb. Eur. J. Immunol. 2009, 39, 2424–2436. [Google Scholar] [CrossRef]
- Kang, S.W.; Lee, S.C.; Park, S.H.; Kim, J.; Kim, H.H.; Lee, H.W.; Seo, S.K.; Kwon, B.S.; Cho, H.R.; Kwon, B. Anti-CD137 suppresses tumor growth by blocking reverse signaling by CD137 ligand. Cancer Res. 2017, 77, 5989–6000. [Google Scholar] [CrossRef] [PubMed]
- De Mingo Pulido, Á.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103+ Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74.e6. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Zhang, L.; Yuan, J.; Babikr, F.; Freywald, A.; Chibbar, R.; Moser, M.; Zhang, W.; Zhang, B.; Fu, Z.; et al. TLR9 agonist enhances radiofrequency ablation-induced CTL responses, leading to the potent inhibition of primary tumor growth and lung metastasis. Cell. Mol. Immunol. 2018. [Google Scholar] [CrossRef]
- Zhuang, Y.; Li, S.; Wang, H.; Pi, J.; Xing, Y.; Li, G. PD-1 blockade enhances radio-immunotherapy efficacy in murine tumor models. J. Cancer Res. Clin. Oncol. 2018, 144, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Den Brok, M.H.; Nierkens, S.; Wagenaars, J.A.; Ruers, T.J.; Schrier, C.C.; Rijke, E.O.; Adema, G.J. Saponin-based adjuvants create a highly effective anti-tumor vaccine when combined with in situ tumor destruction. Vaccine 2012, 30, 737–744. [Google Scholar] [CrossRef]
- Marty-Roix, R.; Vladimer, G.I.; Pouliot, K.; Weng, D.; Buglione-Corbett, R.; West, K.; MacMicking, J.D.; Chee, J.D.; Wang, S.; Lu, S.; et al. Identification of QS-21 as an Inflammasome-activating Molecular Component of Saponin Adjuvants. J. Biol. Chem. 2016, 291, 1123–1136. [Google Scholar] [CrossRef]
- Duewell, P.; Kisser, U.; Heckelsmiller, K.; Hoves, S.; Stoitzner, P.; Koernig, S.; Morelli, A.B.; Clausen, B.E.; Dauer, M.; Eigler, A.; et al. ISCOMATRIX Adjuvant Combines Immune Activation with Antigen Delivery to Dendritic Cells In Vivo Leading to Effective Cross-Priming of CD8+ T Cells. J. Immunol. 2011, 187, 55–63. [Google Scholar] [CrossRef]
- Lu, L.; Yan, H.; Shyam-Sundar, V.; Janowitz, T. Cross-sectional and longitudinal analysis of cancer vaccination trials registered on the US Clinical Trials Database demonstrates paucity of immunological trial endpoints and decline in registration since 2008. Drug Des. Devel. Ther. 2014, 8, 1539–1553. [Google Scholar]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Rodriguez, J.; Castañón, E.; Perez-Gracia, J.L.; Rodriguez, I.; Viudez, A.; Alfaro, C.; Oñate, C.; Perez, G.; Rotellar, F.; Inogés, S.; et al. A randomized phase II clinical trial of dendritic cell vaccination following complete resection of colon cancer liver metastasis. J. Immunother. Cancer 2018, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Matsui, Y.; Ishikawa, A.; Nakajima, T.; Iizasa, T. Randomized controlled phase III trial of adjuvant chemoimmunotherapy with activated cytotoxic T cells and dendritic cells from regional lymph nodes of patients with lung cancer. Cancer Immunol. Immunother. 2018, 67, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Bae, Y.-S. Dendritic cell-based therapeutic cancer vaccines: Past, present and future. Clin. Exp. Vaccine Res. 2014, 3, 113–116. [Google Scholar] [CrossRef]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10, eaao5931. [Google Scholar] [CrossRef]
- Chiang, C.L.-L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Smith, L.; Nisenbaum, H.L.; et al. A Dendritic Cell Vaccine Pulsed with Autologous Hypochlorous Acid-Oxidized Ovarian Cancer Lysate Primes Effective Broad Antitumor Immunity: From Bench to Bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef]
- Joseph, C.G.; Darrah, E.; Shah, A.A.; Skora, A.D.; Casciola-Rosen, L.A.; Wigley, F.M.; Boin, F.; Fava, A.; Thoburn, C.; Kinde, I.; et al. Association of the Autoimmune Disease Scleroderma with an Immunologic Response to Cancer. Science 2014, 343, 152–157. [Google Scholar] [CrossRef]
- Francian, A.; Namen, S.; Stanley, M.; Mann, K.; Martinson, H.; Kullberg, M. Intratumoral delivery of antigen with complement C3-bound liposomes reduces tumor growth in mice. Nanomed. Nanotechnol. Biol. Med. 2018. [Google Scholar] [CrossRef]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.J.G.; Duiveman-de Boer, T.; van de Rakt, M.W.M.M.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clin. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Saung, M.T.; Muth, S.; Ding, D.; Thomas, D.L.; Blair, A.B.; Tsujikawa, T.; Coussens, L.; Jaffee, E.M.; Zheng, L.; Zheng, L. Targeting myeloid-inflamed tumor with anti-CSF-1R antibody expands CD137+ effector T-cells in the murine model of pancreatic cancer. J. Immunother. cancer 2018, 6, 118. [Google Scholar] [CrossRef]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 2019, 177, P556–P571. [Google Scholar] [CrossRef] [PubMed]
- Vo, M.-C.; Nguyen-Pham, T.-N.; Lee, H.-J.; Lakshmi, T.J.; Yang, S.; Jung, S.-H.; Kim, H.-J.; Lee, J.-J. Combination therapy with dendritic cells and lenalidomide is an effective approach to enhance antitumor immunity in a mouse colon cancer model. Oncotarget 2017, 8, 27252–27262. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef]


{kind=link}
| ClinicalTrials.Gov Identifier | Title of Clinical Trial | DC Used | Antigen Loaded | Concurrent Treatments | Treated Cancer | Phase |
|---|---|---|---|---|---|---|
| NCT03410732 | Dendritic Cell-based Immunotherapy in Treatment Gastric Cancer | Mo-DC | proteins from autologous tumour cell membrane | Post-radical surgery | Gastric Cancer | 2 |
| NCT03406715 | Combination Immunotherapy-Ipilimumab-Nivolumab-Dendritic Cell p53 Vac-Patients With Small Cell Lung Cancer (SCLC) | Mo-DC | p53 | Ipilimumab, Nivolumab | Small Cell Lung Cancer | 2 |
| NCT00703105 | Ovarian Dendritic Cell Vaccine Trial | Mo-DC | Tumour lysate | ONTAK | Ovarian Cancer | 2 |
| NCT02033616 | Autologous Dendritic Cells Loaded With Autologous Tumor Associated Antigens for Treatment of Advanced Epithelial Ovarian Carcinomas | Mo-DC | Autologous tumour associated antigens | - | Ovarian, fallopian and peritoneal cancer | 2 |
| NCT02718391 | Complementary Vaccination With Dendritic Cells Pulsed With Autologous Tumor Lysate in Resected Stage III and IV Melanoma Patients. (ACDC) | Mo-DC | autologous tumour lysate or homogenate | post-surgical resection, IL-2 | Melanoma | 2 |
| NCT01686334 | Efficacy Study of Dendritic Cell Vaccination in Patients With Acute Myeloid Leukemia in Remission (WIDEA) | Mo-DC | Wilms’ tumour antigen | - | Acute Myeloid Leukemia | 1, 2 |
| NCT03610360 | DENdritic Cell Immunotherapy for Mesothelioma (DENIM) | Mo-DC | Allogenic tumour cell lysate | - | Mesothelioma | 2 |
| NCT03697707 | Efficacy and Safety of Immunotherapy With Allogeneic Dendritic Cells, DCP-001, in Patients With Acute Myeloid Leukaemia (ADVANCE-II) | Mo-DC | DCP-001 | - | Acute Myeloid Leukaemia | 2 |
| NCT00338377 | Lymphodepletion Plus Adoptive Cell Transfer With or Without Dendritic Cell Immunization in Patients With Metastatic Melanoma | Mo-DC | MART-1 | Cyclophosphamide, Fludarabine, IL-2, T cells, Mesna | Melanoma | 2 |
| NCT02649582 | Adjuvant Dendritic Cell-immunotherapy Plus Temozolomide in Glioblastoma Patients (ADDIT-GLIO) | Mo-DC | Wilms’ tumour antigen | Temozolmide | Glioblastoma | 2 |
| NCT03325101 | Dendritic Cell Therapy After Cryosurgery in Combination With Pembrolizumab in Treating Patients With Stage III-IV Melanoma That Cannot Be Remove by Surgery | Mo-DC | - | Pembrolizumab | Melanoma | 1b, 2 |
| NCT02334735 | A Comparison of Matured Dendritic Cells and Montanide® in Study Subjects With High Risk of Melanoma Recurrence | Mo-DC | KLH, NY-ESO-1, MART-1 | Poly-ICLC | Melanoma | 2 |
| NCT02709616 | Personalized Cellular Vaccine for Glioblastoma (PERCELLVAC) (PERCELLVAC) | Mo-DC | tumour antigen mRNA | - | Glioblastoma | 1, 2 |
| NCT01976585 | In Situ Vaccine for Low-Grade Lymphoma: Combination of Intratumoral Flt3L and Poly-ICLC With Low-Dose Radiotherapy | Tumoural DCs | - | Flt3L, Poly-ICLC | Lymphoma | 2 |
| NCT01983748 | Dendritic Cells Plus Autologous Tumor RNA in Uveal Melanoma | Mo-DC | Autologous tumour RNA | - | Uveal melanoma | 3 |
| NCT02993315 | Melanoma Patients Immunized With Natural DenDritic Cells (MIND-DC) | mDC and pDC | antigen-loaded | - | Melanoma | 3, 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murgaski, A.; Bardet, P.M.R.; Arnouk, S.M.; Clappaert, E.J.; Laoui, D. Unleashing Tumour-Dendritic Cells to Fight Cancer by Tackling Their Three A’s: Abundance, Activation and Antigen-Delivery. Cancers 2019, 11, 670. https://doi.org/10.3390/cancers11050670
Murgaski A, Bardet PMR, Arnouk SM, Clappaert EJ, Laoui D. Unleashing Tumour-Dendritic Cells to Fight Cancer by Tackling Their Three A’s: Abundance, Activation and Antigen-Delivery. Cancers. 2019; 11(5):670. https://doi.org/10.3390/cancers11050670
Chicago/Turabian StyleMurgaski, Aleksandar, Pauline M. R. Bardet, Sana M. Arnouk, Emile J. Clappaert, and Damya Laoui. 2019. "Unleashing Tumour-Dendritic Cells to Fight Cancer by Tackling Their Three A’s: Abundance, Activation and Antigen-Delivery" Cancers 11, no. 5: 670. https://doi.org/10.3390/cancers11050670
APA StyleMurgaski, A., Bardet, P. M. R., Arnouk, S. M., Clappaert, E. J., & Laoui, D. (2019). Unleashing Tumour-Dendritic Cells to Fight Cancer by Tackling Their Three A’s: Abundance, Activation and Antigen-Delivery. Cancers, 11(5), 670. https://doi.org/10.3390/cancers11050670