Perturbing Enhancer Activity in Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
Abstract
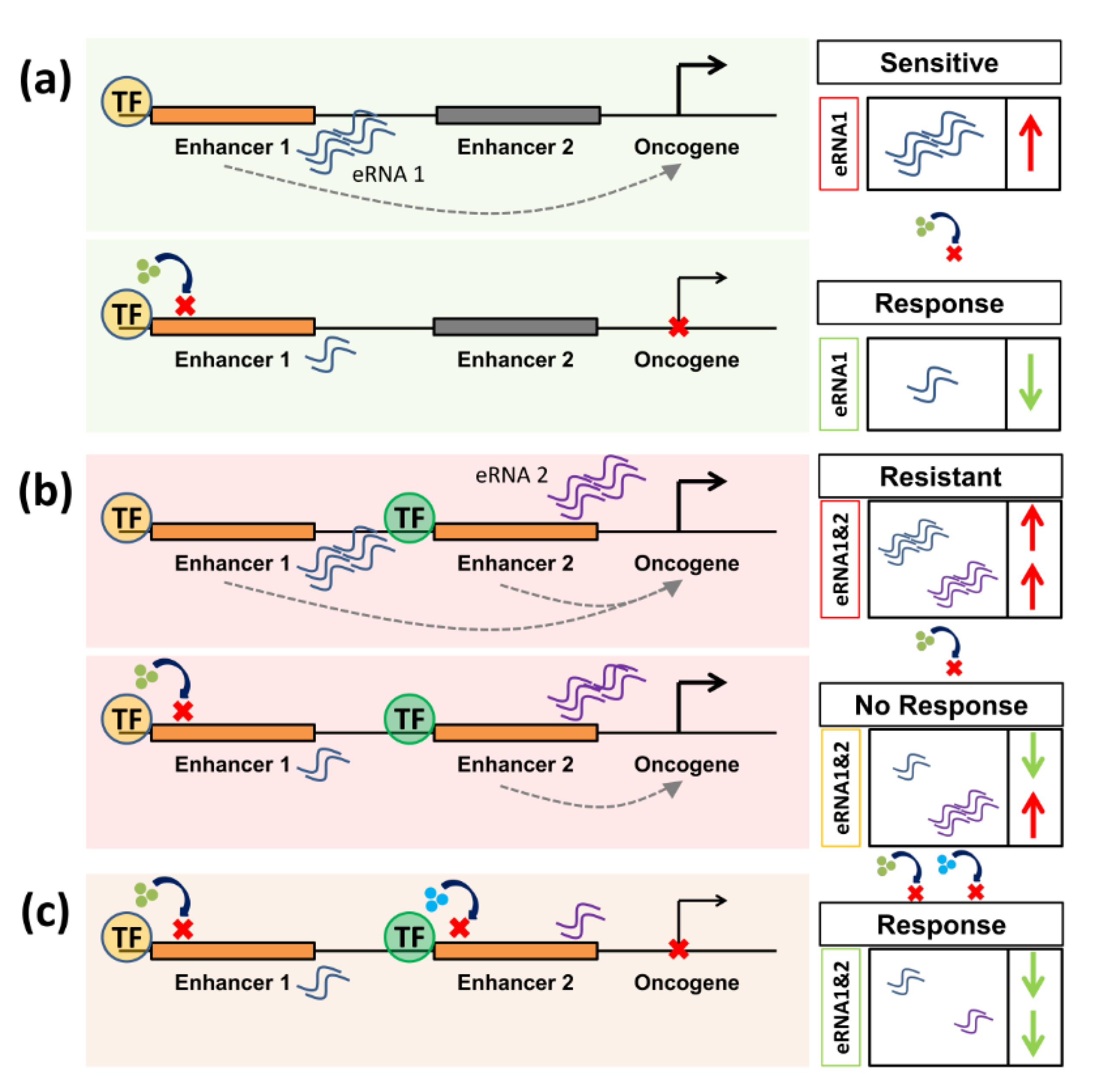
1. Introduction
2. Targeting Transcription Factor-Related Programs in Cancer
3. Perturbing Enhancer Activity by Therapeutic Agents and Inhibitors
3.1. Enhancers Are Context-Specific and Indispensable for Cancer Cells
3.2. Activity of Enhancers Can Be Pharmacologically Perturbed
3.2.1. Epigenetic Modulators
3.2.2. Cyclin-Dependent Kinase Inhibitors
4. Challenges Facing the Utility of Enhancer Modulators in the Clinical Setting
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Suzuki, A.; Makinoshima, H.; Wakaguri, H.; Esumi, H.; Sugano, S.; Kohno, T.; Tsuchihara, K.; Suzuki, Y. Aberrant transcriptional regulations in cancers: Genome, transcriptome and epigenome analysis of lung adenocarcinoma cell lines. Nucleic Acids Res. 2014, 42, 13557–13572. [Google Scholar] [CrossRef]
- Lu, B.; Klingbeil, O.; Tarumoto, Y.; Somerville, T.D.D.; Huang, Y.H.; Wei, Y.; Wai, D.C.; Low, J.K.K.; Milazzo, J.P.; Wu, X.S.; et al. A Transcription Factor Addiction in Leukemia Imposed by the MLL Promoter Sequence. Cancer Cell 2018, 34, 970–981.e8. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Toohey, M.G.; Morley, K.L.; Peterson, D.O. Multiple hormone-inducible enhancers as mediators of differential transcription. Mol. Cell. Biol. 1986, 6, 4526–4538. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Vahedi, G.; Kanno, Y.; Furumoto, Y.; Jiang, K.; Parker, S.C.; Erdos, M.R.; Davis, S.R.; Roychoudhuri, R.; Restifo, N.P.; Gadina, M.; et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 2015, 520, 558–562. [Google Scholar] [CrossRef]
- Sun, W.; Yao, S.; Tang, J.; Liu, S.; Chen, J.; Deng, D.; Zeng, C. Integrative analysis of super enhancer SNPs for type 2 diabetes. PLoS ONE 2018, 13, e0192105. [Google Scholar] [CrossRef]
- Groschel, S.; Sanders, M.A.; Hoogenboezem, R.; de Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; van der Velden, V.H.J.; Havermans, M.; Avellino, R.; van Lom, K.; et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef]
- Takeda, D.Y.; Spisak, S.; Seo, J.H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szallasi, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432.e3. [Google Scholar] [CrossRef]
- Zeid, R.; Lawlor, M.A.; Poon, E.; Reyes, J.M.; Fulciniti, M.; Lopez, M.A.; Scott, T.G.; Nabet, B.; Erb, M.A.; Winter, G.E.; et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 2018, 50, 515–523. [Google Scholar] [CrossRef]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stutz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20. [Google Scholar] [CrossRef]
- Somerville, T.D.D.; Xu, Y.; Miyabayashi, K.; Tiriac, H.; Cleary, C.R.; Maia-Silva, D.; Milazzo, J.P.; Tuveson, D.A.; Vakoc, C.R. TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep. 2018, 25, 1741–1755.e7. [Google Scholar] [CrossRef]
- Hamdan, F.H.; Johnsen, S.A. DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc. Natl. Acad. Sci. USA 2018, 115, E12343–E12352. [Google Scholar] [CrossRef]
- Knoechel, B.; Roderick, J.E.; Williamson, K.E.; Zhu, J.; Lohr, J.G.; Cotton, M.J.; Gillespie, S.M.; Fernandez, D.; Ku, M.; Wang, H.; et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 364–370. [Google Scholar] [CrossRef]
- Smith, E.; Shilatifard, A. Enhancer biology and enhanceropathies. Nat. Struct. Mol. Biol. 2014, 21, 210–219. [Google Scholar] [CrossRef]
- Palstra, R.J.; Grosveld, F. Transcription factor binding at enhancers: Shaping a genomic regulatory landscape in flux. Front. Genet. 2012, 3, 195. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef]
- Najafova, Z.; Tirado-Magallanes, R.; Subramaniam, M.; Hossan, T.; Schmidt, G.; Nagarajan, S.; Baumgart, S.J.; Mishra, V.K.; Bedi, U.; Hesse, E.; et al. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2017, 45, 127–141. [Google Scholar] [CrossRef]
- Lan, X.; Witt, H.; Katsumura, K.; Ye, Z.; Wang, Q.; Bresnick, E.H.; Farnham, P.J.; Jin, V.X. Integration of Hi-C and ChIP-seq data reveals distinct types of chromatin linkages. Nucleic Acids Res. 2012, 40, 7690–7704. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nature Med. 2019. [Google Scholar] [CrossRef]
- Tsang, F.H.; Law, C.T.; Tang, T.C.; Cheng, C.L.; Chin, D.W.; Tam, W.V.; Wei, L.; Wong, C.C.; Ng, I.O.; Wong, C.M. Aberrant Super-Enhancer Landscape in Human Hepatocellular Carcinoma. Hepatology 2019. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Xu, X.; Gao, J.; Dai, W.; Wang, D.; Wu, J.; Wang, J. Gene activation by a CRISPR-assisted trans enhancer. eLife 2019, 8, e45973. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, K.; Wang, C.; Zhang, Z.; Zheng, C.; Zhao, Y.; Zheng, Y.; Liu, C.; An, Y.; Shi, L.; et al. Multistage Delivery Nanoparticle Facilitates Efficient CRISPR/dCas9 Activation and Tumor Growth Suppression In Vivo. Adv. Sci. (Weinh) 2018, 6, 1801423. [Google Scholar] [CrossRef]
- Ward, H.W. Anti-oestrogen therapy for breast cancer: A trial of tamoxifen at two dose levels. Br. Med. J. 1973, 1, 13–14. [Google Scholar] [CrossRef]
- Moseson, D.L.; Sasaki, G.H.; Kraybill, W.G.; Leung, B.S.; Davenport, C.E.; Fletcher, W.S. The use of antiestrogens tamoxifen and nafoxidine in the treatment of human breast cancer in correlation with estrogen receptor values. A phase II study. Cancer 1978, 41, 797–802. [Google Scholar] [CrossRef]
- Manni, A.; Trujillo, J.E.; Marshall, J.S.; Brodkey, J.; Pearson, O.H. Antihormone treatment of stage IV breast cancer. Cancer 1979, 43, 444–450. [Google Scholar] [CrossRef]
- Labrie, F.; Dupont, A.; Belanger, A.; Lefebvre, F.A.; Cusan, L.; Raynaud, J.P.; Husson, J.M.; Fazekas, A.T. New hormonal therapy in prostate cancer: Combined use of a pure antiandrogen and an LHRH agonist. Horm. Res. 1983, 18, 18–27. [Google Scholar] [CrossRef]
- Moguilewsky, M.; Fiet, J.; Tournemine, C.; Raynaud, J.P. Pharmacology of an antiandrogen, anandron, used as an adjuvant therapy in the treatment of prostate cancer. J. Steroid Biochem. 1986, 24, 139–146. [Google Scholar] [CrossRef]
- Soloway, M.S. Newer methods of hormonal therapy for prostate cancer. Urology 1984, 24, 30–38. [Google Scholar]
- Yamamoto, K.R.; Stampfer, M.R.; Tomkins, G.M. Receptors from glucocorticoid-sensitive lymphoma cells and two clases of insensitive clones: Physical and DNA-binding properties. Proc. Natl. Acad. Sci. USA 1974, 71, 3901–3905. [Google Scholar] [CrossRef]
- Gehring, U.; Mohit, B.; Tomkins, G.M. Glucocorticoid action on hybrid clones derived from cultured myeloma and lymphoma cell lines. Proc. Natl. Acad. Sci. USA 1972, 69, 3124–3127. [Google Scholar] [CrossRef]
- Lumachi, F.; Brunello, A.; Maruzzo, M.; Basso, U.; Basso, S.M. Treatment of estrogen receptor-positive breast cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef]
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Carroll, J.S.; Meyer, C.A.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006, 38, 1289–1297. [Google Scholar] [CrossRef]
- Nair, S.J.; Yang, L.; Meluzzi, D.; Oh, S.; Yang, F.; Friedman, M.J.; Wang, S.; Suter, T.; Alshareedah, I.; Gamliel, A.; et al. Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat. Struct. Mol. Biol. 2019, 26, 193–203. [Google Scholar] [CrossRef]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef]
- Makkonen, H.; Kauhanen, M.; Paakinaho, V.; Jaaskelainen, T.; Palvimo, J.J. Long-range activation of FKBP51 transcription by the androgen receptor via distal intronic enhancers. Nucleic Acids Res. 2009, 37, 4135–4148. [Google Scholar] [CrossRef]
- Wang, D.; Garcia-Bassets, I.; Benner, C.; Li, W.; Su, X.; Zhou, Y.; Qiu, J.; Liu, W.; Kaikkonen, M.U.; Ohgi, K.A.; et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011, 474, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Pihlajamaa, P.; Sahu, B.; Lyly, L.; Aittomaki, V.; Hautaniemi, S.; Janne, O.A. Tissue-specific pioneer factors associate with androgen receptor cistromes and transcription programs. EMBO J. 2014, 33, 312–326. [Google Scholar] [CrossRef] [PubMed]
- McDowell, I.C.; Barrera, A.; D’Ippolito, A.M.; Vockley, C.M.; Hong, L.K.; Leichter, S.M.; Bartelt, L.C.; Majoros, W.H.; Song, L.; Safi, A.; et al. Glucocorticoid receptor recruits to enhancers and drives activation by motif-directed binding. Genome Res. 2018, 28, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Quang, C.T.; Leboucher, S.; Passaro, D.; Fuhrmann, L.; Nourieh, M.; Vincent-Salomon, A.; Ghysdael, J. The calcineurin/NFAT pathway is activated in diagnostic breast cancer cases and is essential to survival and metastasis of mammary cancer cells. Cell Death Dis. 2015, 6, e1658. [Google Scholar] [CrossRef]
- Takashima, T.; Fujiwara, Y.; Higuchi, K.; Arakawa, T.; Yano, Y.; Hasuma, T.; Otani, S. PPAR-gamma ligands inhibit growth of human esophageal adenocarcinoma cells through induction of apoptosis, cell cycle arrest and reduction of ornithine decarboxylase activity. Int. J. Oncol. 2001, 19, 465–471. [Google Scholar] [PubMed]
- Fujii, D.; Yoshida, K.; Tanabe, K.; Hihara, J.; Toge, T. The ligands of peroxisome proliferator-activated receptor (PPAR) gamma inhibit growth of human esophageal carcinoma cells through induction of apoptosis and cell cycle arrest. Anticancer Res. 2004, 24, 1409–1416. [Google Scholar] [PubMed]
- Motomura, W.; Takahashi, N.; Nagamine, M.; Sawamukai, M.; Tanno, S.; Kohgo, Y.; Okumura, T. Growth arrest by troglitazone is mediated by p27Kip1 accumulation, which results from dual inhibition of proteasome activity and Skp2 expression in human hepatocellular carcinoma cells. Int. J. Cancer 2004, 108, 41–46. [Google Scholar] [CrossRef]
- Liao, K.F.; Lin, C.L.; Lai, S.W. Association between colorectal cancer and thiazolidinediones administration in a case-control study. BioMedicine 2019, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the NFAT family: Regulation and function. Annu. Rev. Immunol. 1997, 15, 707–747. [Google Scholar] [CrossRef]
- Wang, S.; Kang, X.; Cao, S.; Cheng, H.; Wang, D.; Geng, J. Calcineurin/NFATc1 pathway contributes to cell proliferation in hepatocellular carcinoma. Dig. Dis. Sci. 2012, 57, 3184–3188. [Google Scholar] [CrossRef] [PubMed]
- Perotti, V.; Baldassari, P.; Bersani, I.; Molla, A.; Vegetti, C.; Tassi, E.; Dal Col, J.; Dolcetti, R.; Anichini, A.; Mortarini, R. NFATc2 is a potential therapeutic target in human melanoma. J. Invest. Dermatol. 2012, 132, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Norrmen, C.; Ivanov, K.I.; Cheng, J.; Zangger, N.; Delorenzi, M.; Jaquet, M.; Miura, N.; Puolakkainen, P.; Horsley, V.; Hu, J.; et al. FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J. Cell Biol. 2009, 185, 439–457. [Google Scholar] [CrossRef]
- Baumgart, S.; Chen, N.M.; Siveke, J.T.; Konig, A.; Zhang, J.S.; Singh, S.K.; Wolf, E.; Bartkuhn, M.; Esposito, I.; Hessmann, E.; et al. Inflammation-induced NFATc1-STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D. Cancer Discov. 2014, 4, 688–701. [Google Scholar] [CrossRef]
- Buchholz, M.; Schatz, A.; Wagner, M.; Michl, P.; Linhart, T.; Adler, G.; Gress, T.M.; Ellenrieder, V. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006, 25, 3714–3724. [Google Scholar] [CrossRef]
- Wu, X.; Nguyen, B.C.; Dziunycz, P.; Chang, S.; Brooks, Y.; Lefort, K.; Hofbauer, G.F.; Dotto, G.P. Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature 2010, 465, 368–372. [Google Scholar] [CrossRef]
- Baba, Y.; Nosho, K.; Shima, K.; Irahara, N.; Chan, A.T.; Meyerhardt, J.A.; Chung, D.C.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. HIF1A overexpression is associated with poor prognosis in a cohort of 731 colorectal cancers. Am. J. Pathol. 2010, 176, 2292–2301. [Google Scholar] [CrossRef]
- Wu, Y.; Yun, D.; Zhao, Y.; Wang, Y.; Sun, R.; Yan, Q.; Zhang, S.; Lu, M.; Zhang, Z.; Lu, D.; et al. Down regulation of RNA binding motif, single-stranded interacting protein 3, along with up regulation of nuclear HIF1A correlates with poor prognosis in patients with gastric cancer. Oncotarget 2017, 8, 1262–1277. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.T.; Chua, M.S.; Whitlock, J.P., Jr.; Shin, Y.C.; Song, W.H.; Kim, Y.; Eom, C.Y.; An, W.G. Rodent-specific hypoxia response elements enhance PAI-1 expression through HIF-1 or HIF-2 in mouse hepatoma cells. Int. J. Oncol. 2010, 37, 1627–1638. [Google Scholar]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef]
- Tausendschon, M.; Rehli, M.; Dehne, N.; Schmidl, C.; Doring, C.; Hansmann, M.L.; Brune, B. Genome-wide identification of hypoxia-inducible factor-1 and -2 binding sites in hypoxic human macrophages alternatively activated by IL-10. Biochim. Biophys. Acta 2015, 1849, 10–22. [Google Scholar] [CrossRef]
- Rapisarda, A.; Uranchimeg, B.; Sordet, O.; Pommier, Y.; Shoemaker, R.H.; Melillo, G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: Mechanism and therapeutic implications. Cancer Res. 2004, 64, 1475–1482. [Google Scholar] [CrossRef]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef]
- Xiong, L.; Kang, R.; Ding, R.; Kang, W.; Zhang, Y.; Liu, W.; Huang, Q.; Meng, J.; Guo, Z. Genome-wide Identification and Characterization of Enhancers Across 10 Human Tissues. Int. J. Biol. Sci. 2018, 14, 1321–1332. [Google Scholar] [CrossRef]
- Cao, Q.; Anyansi, C.; Hu, X.; Xu, L.; Xiong, L.; Tang, W.; Mok, M.T.S.; Cheng, C.; Fan, X.; Gerstein, M.; et al. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet. 2017, 49, 1428–1436. [Google Scholar] [CrossRef]
- He, B.; Chen, C.; Teng, L.; Tan, K. Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2191–E2199. [Google Scholar] [CrossRef]
- Hu, C.Y.; Mohtat, D.; Yu, Y.; Ko, Y.A.; Shenoy, N.; Bhattacharya, S.; Izquierdo, M.C.; Park, A.S.; Giricz, O.; Vallumsetla, N.; et al. Kidney cancer is characterized by aberrant methylation of tissue-specific enhancers that are prognostic for overall survival. Clin. Cancer Res. 2014, 20, 4349–4360. [Google Scholar] [CrossRef]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018, 33, 512–526.e8. [Google Scholar] [CrossRef]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Zhu, H.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef]
- Zawistowski, J.S.; Bevill, S.M.; Goulet, D.R.; Stuhlmiller, T.J.; Beltran, A.S.; Olivares-Quintero, J.F.; Singh, D.; Sciaky, N.; Parker, J.S.; Rashid, N.U.; et al. Enhancer Remodeling during Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacologic Targeting of the P-TEFb Complex. Cancer Discov. 2017, 7, 302–321. [Google Scholar] [CrossRef]
- Nagarajan, S.; Hossan, T.; Alawi, M.; Najafova, Z.; Indenbirken, D.; Bedi, U.; Taipaleenmaki, H.; Ben-Batalla, I.; Scheller, M.; Loges, S.; et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep. 2014, 8, 460–469. [Google Scholar] [CrossRef]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nature Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef]
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.S.; et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953. [Google Scholar] [CrossRef]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Lee, J.S.; Smith, E.; Shilatifard, A. The language of histone crosstalk. Cell 2010, 142, 682–685. [Google Scholar] [CrossRef]
- Musselman, C.A.; Lalonde, M.E.; Cote, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef] [PubMed]
- Mirguet, O.; Lamotte, Y.; Donche, F.; Toum, J.; Gellibert, F.; Bouillot, A.; Gosmini, R.; Nguyen, V.L.; Delannee, D.; Seal, J.; et al. From ApoA1 upregulation to BET family bromodomain inhibition: Discovery of I-BET151. Bioorg. Med. Chem. Lett. 2012, 22, 2963–2967. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Imielinski, M.; Watanabe, H.; Cherniack, A.D.; Meyerson, M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet. 2016, 48, 176–182. [Google Scholar] [CrossRef]
- Bojcsuk, D.; Nagy, G.; Balint, B.L. Inducible super-enhancers are organized based on canonical signal-specific transcription factor binding elements. Nucleic Acids Res. 2017, 45, 3693–3706. [Google Scholar] [CrossRef]
- Gerard, D.; Schmidt, F.; Ginolhac, A.; Schmitz, M.; Halder, R.; Ebert, P.; Schulz, M.H.; Sauter, T.; Sinkkonen, L. Temporal enhancer profiling of parallel lineages identifies AHR and GLIS1 as regulators of mesenchymal multipotency. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Saiakhova, A.; Corradin, O.; Luppino, J.M.; Lovrenert, K.; Bartels, C.F.; Morrow, J.J.; Mack, S.C.; Dhillon, G.; Beard, L.; et al. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat. Commun. 2017, 8, 14400. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2018, 553, 101–105. [Google Scholar] [CrossRef]
- Ott, C.J.; Federation, A.J.; Schwartz, L.S.; Kasar, S.; Klitgaard, J.L.; Lenci, R.; Li, Q.; Lawlor, M.; Fernandes, S.M.; Souza, A.; et al. Enhancer Architecture and Essential Core Regulatory Circuitry of Chronic Lymphocytic Leukemia. Cancer Cell 2018, 34, 982–995.e7. [Google Scholar] [CrossRef]
- Hemming, M.L.; Lawlor, M.A.; Andersen, J.L.; Hagan, T.; Chipashvili, O.; Scott, T.G.; Raut, C.P.; Sicinska, E.; Armstrong, S.A.; Demetri, G.D.; et al. Enhancer Domains in Gastrointestinal Stromal Tumor Regulate KIT Expression and Are Targetable by BET Bromodomain Inhibition. Cancer Res. 2019, 79, 994–1009. [Google Scholar] [CrossRef]
- Xu, Y.M.; Du, J.Y.; Lau, A.T. Posttranslational modifications of human histone H3: An update. Proteomics 2014, 14, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Willson, T.A.; Wakefield, M.J.; Trounson, E.; Hilton, D.J.; Blewitt, M.E.; Oshlack, A.; Majewski, I.J. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011, 39, 7415–7427. [Google Scholar] [CrossRef]
- Arthur, R.K.; Ma, L.; Slattery, M.; Spokony, R.F.; Ostapenko, A.; Negre, N.; White, K.P. Evolution of H3K27me3-marked chromatin is linked to gene expression evolution and to patterns of gene duplication and diversification. Genome Res. 2014, 24, 1115–1124. [Google Scholar] [CrossRef]
- Xu, B.; On, D.M.; Ma, A.; Parton, T.; Konze, K.D.; Pattenden, S.G.; Allison, D.F.; Cai, L.; Rockowitz, S.; Liu, S.; et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 2015, 125, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.P.; Ling, K. EZH2 and histone deacetylase inhibitors induce apoptosis in triple negative breast cancer cells by differentially increasing H3 Lys(27) acetylation in the BIM gene promoter and enhancers. Oncol. Lett. 2017, 14, 5735–5742. [Google Scholar] [CrossRef] [PubMed]
- Torres, I.O.; Fujimori, D.G. Functional coupling between writers, erasers and readers of histone and DNA methylation. Curr. Opin. Struct. Biol. 2015, 35, 68–75. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Sanchez, G.J.; Richmond, P.A.; Bunker, E.N.; Karman, S.S.; Azofeifa, J.; Garnett, A.T.; Xu, Q.; Wheeler, G.E.; Toomey, C.M.; Zhang, Q.; et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2018, 46, 1756–1776. [Google Scholar] [CrossRef]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef]
- Sawan, C.; Herceg, Z. Histone modifications and cancer. Adv. Genet. 2010, 70, 57–85. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Maiques-Diaz, A.; Spencer, G.J.; Lynch, J.T.; Ciceri, F.; Williams, E.L.; Amaral, F.M.R.; Wiseman, D.H.; Harris, W.J.; Li, Y.; Sahoo, S.; et al. Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep. 2018, 22, 3641–3659. [Google Scholar] [CrossRef]
- Whyte, W.A.; Bilodeau, S.; Orlando, D.A.; Hoke, H.A.; Frampton, G.M.; Foster, C.T.; Cowley, S.M.; Young, R.A. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 2012, 482, 221–225. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Metzger, E.; Imhof, A.; Patel, D.; Kahl, P.; Hoffmeyer, K.; Friedrichs, N.; Muller, J.M.; Greschik, H.; Kirfel, J.; Ji, S.; et al. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature 2010, 464, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Kwon, Y.S.; Nunez, E.; Cardamone, M.D.; Hutt, K.R.; Ohgi, K.A.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G.; et al. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc. Natl. Acad. Sci. USA 2008, 105, 19199–19204. [Google Scholar] [CrossRef] [PubMed]
- Buratowski, S. Progression through the RNA polymerase II CTD cycle. Mol. Cell 2009, 36, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; LeRoy, G.; Campos, E.; Sun, H.W.; Brooks, S.R.; Vahedi, G.; Heightman, T.D.; Garcia, B.A.; Reinberg, D.; et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014, 21, 1047–1057. [Google Scholar] [CrossRef]
- Scheidegger, A.; Nechaev, S. RNA polymerase II pausing as a context-dependent reader of the genome. Biochem. Cell Biol. 2016, 94, 82–92. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Shibata, H.; Handa, H. Transcription elongation factors DSIF and NELF: Promoter-proximal pausing and beyond. Biochim. Biophys. Acta 2013, 1829, 98–104. [Google Scholar] [CrossRef]
- Kwak, H.; Lis, J.T. Control of transcriptional elongation. Ann. Rev. Genet. 2013, 47, 483–508. [Google Scholar] [CrossRef]
- Nechaev, S.; Adelman, K. Pol II waiting in the starting gates: Regulating the transition from transcription initiation into productive elongation. Biochim. Biophys. Acta 2011, 1809, 34–45. [Google Scholar] [CrossRef]
- Marshall, N.F.; Price, D.H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef]
- Liu, X.; Kraus, W.L.; Bai, X. Ready, pause, go: Regulation of RNA polymerase II pausing and release by cellular signaling pathways. Trends Biochem. Sci. 2015, 40, 516–525. [Google Scholar] [CrossRef]
- Washburn, R.S.; Gottesman, M.E. Regulation of transcription elongation and termination. Biomolecules 2015, 5, 1063–1078. [Google Scholar] [CrossRef]
- Hah, N.; Murakami, S.; Nagari, A.; Danko, C.G.; Kraus, W.L. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013, 23, 1210–1223. [Google Scholar] [CrossRef]
- Gerlach, D.; Tontsch-Grunt, U.; Baum, A.; Popow, J.; Scharn, D.; Hofmann, M.H.; Engelhardt, H.; Kaya, O.; Beck, J.; Schweifer, N.; et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene 2018, 37, 2687–2701. [Google Scholar] [CrossRef]
- Moreno, N.; Holsten, T.; Mertins, J.; Zhogbi, A.; Johann, P.; Kool, M.; Meisterernst, M.; Kerl, K. Combined BRD4 and CDK9 inhibition as a new therapeutic approach in malignant rhabdoid tumors. Oncotarget 2017, 8, 84986–84995. [Google Scholar] [CrossRef]
- Boffo, S.; Damato, A.; Alfano, L.; Giordano, A. CDK9 inhibitors in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 36. [Google Scholar] [CrossRef]
- Sharifnia, T.; Wawer, M.J.; Chen, T.; Huang, Q.Y.; Weir, B.A.; Sizemore, A.; Lawlor, M.A.; Goodale, A.; Cowley, G.S.; Vazquez, F.; et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nature Med. 2019, 25, 292–300. [Google Scholar] [CrossRef]
- Fisher, R.P.; Morgan, D.O. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 1994, 78, 713–724. [Google Scholar] [CrossRef]
- Schwartz, B.E.; Larochelle, S.; Suter, B.; Lis, J.T. Cdk7 is required for full activation of Drosophila heat shock genes and RNA polymerase II phosphorylation in vivo. Mol. Cell. Biol. 2003, 23, 6876–6886. [Google Scholar] [CrossRef]
- Lolli, G. Binding to DNA of the RNA-polymerase II C-terminal domain allows discrimination between Cdk7 and Cdk9 phosphorylation. Nucleic Acids Res. 2009, 37, 1260–1268. [Google Scholar] [CrossRef]
- Larochelle, S.; Merrick, K.A.; Terret, M.E.; Wohlbold, L.; Barboza, N.M.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V.; Fisher, R.P. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol. Cell 2007, 25, 839–850. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef]
- Zhang, Z.; Peng, H.; Wang, X.; Yin, X.; Ma, P.; Jing, Y.; Cai, M.C.; Liu, J.; Zhang, M.; Zhang, S.; et al. Preclinical Efficacy and Molecular Mechanism of Targeting CDK7-Dependent Transcriptional Addiction in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 1739–1750. [Google Scholar] [CrossRef]
- Jiang, Y.Y.; Lin, D.C.; Mayakonda, A.; Hazawa, M.; Ding, L.W.; Chien, W.W.; Xu, L.; Chen, Y.; Xiao, J.F.; Senapedis, W.; et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut 2017, 66, 1358–1368. [Google Scholar] [CrossRef]
- Eliades, P.; Abraham, B.J.; Ji, Z.; Miller, D.M.; Christensen, C.L.; Kwiatkowski, N.; Kumar, R.; Njauw, C.N.; Taylor, M.; Miao, B.; et al. High MITF Expression Is Associated with Super-Enhancers and Suppressed by CDK7 Inhibition in Melanoma. J. Invest. Dermatol. 2018, 138, 1582–1590. [Google Scholar] [CrossRef]
- Meng, W.; Wang, J.; Wang, B.; Liu, F.; Li, M.; Zhao, Y.; Zhang, C.; Li, Q.; Chen, J.; Zhang, L.; et al. CDK7 inhibition is a novel therapeutic strategy against GBM both in vitro and in vivo. Cancer Manag. Res. 2018, 10, 5747–5758. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Geng, J.; Zhang, L.; Wang, Y.; Niu, N.; Fang, Y.; Liu, F.; Shi, J.; Zhang, Z.G.; Sun, Y.W.; et al. THZ1 reveals CDK7-dependent transcriptional addictions in pancreatic cancer. Oncogene 2019. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, L.; Jiang, G.; Chen, Z.; Li, J.; An, P.; Chen, L.; Du, J.; Wang, H. Targeting CDK7 increases the stability of Snail to promote the dissemination of colorectal cancer. Cell Death Differ. 2018. [Google Scholar] [CrossRef]
- Nilson, K.A.; Guo, J.; Turek, M.E.; Brogie, J.E.; Delaney, E.; Luse, D.S.; Price, D.H. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol. Cell 2015, 59, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Sampathi, S.; Acharya, P.; Zhao, Y.; Wang, J.; Stengel, K.R.; Liu, Q.; Savona, M.R.; Hiebert, S.W. The CDK7 inhibitor THZ1 alters RNA polymerase dynamics at the 5′ and 3′ ends of genes. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Olson, C.M.; Liang, Y.; Leggett, A.; Park, W.D.; Li, L.; Mills, C.E.; Elsarrag, S.Z.; Ficarro, S.B.; Zhang, T.; Duster, R.; et al. Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype. Cell Chem. Biol. 2019. [Google Scholar] [CrossRef]
- Kornberg, R.D. Mediator and the mechanism of transcriptional activation. Trends Biochem. Sci. 2005, 30, 235–239. [Google Scholar] [CrossRef]
- Poss, Z.C.; Ebmeier, C.C.; Taatjes, D.J. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 575–608. [Google Scholar] [CrossRef]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Dunn, T.M.; Hahn, S.; Ogden, S.; Schleif, R.F. An operator at -280 base pairs that is required for repression of araBAD operon promoter: Addition of DNA helical turns between the operator and promoter cyclically hinders repression. Proc. Natl. Acad. Sci. USA 1984, 81, 5017–5020. [Google Scholar] [CrossRef]
- Wendt, K.S.; Yoshida, K.; Itoh, T.; Bando, M.; Koch, B.; Schirghuber, E.; Tsutsumi, S.; Nagae, G.; Ishihara, K.; Mishiro, T.; et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008, 451, 796–801. [Google Scholar] [CrossRef]
- Bell, A.C.; West, A.G.; Felsenfeld, G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 1999, 98, 387–396. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Z.; Bando, M.; Itoh, T.; Deardorff, M.A.; Clark, D.; Kaur, M.; Tandy, S.; Kondoh, T.; Rappaport, E.; et al. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 2009, 7, e1000119. [Google Scholar] [CrossRef]
- Lewis, B.A.; Reinberg, D. The mediator coactivator complex: Functional and physical roles in transcriptional regulation. J. Cell Sci. 2003, 116, 3667–3675. [Google Scholar] [CrossRef]
- Petrenko, N.; Jin, Y.; Wong, K.H.; Struhl, K. Mediator Undergoes a Compositional Change during Transcriptional Activation. Mol. Cell 2016, 64, 443–454. [Google Scholar] [CrossRef]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef]
- Pawar, A.; Gollavilli, P.N.; Wang, S.; Asangani, I.A. Resistance to BET Inhibitor Leads to Alternative Therapeutic Vulnerabilities in Castration-Resistant Prostate Cancer. Cell Rep. 2018, 22, 2236–2245. [Google Scholar] [CrossRef]
- Kumar, K.; Raza, S.S.; Knab, L.M.; Chow, C.R.; Kwok, B.; Bentrem, D.J.; Popovic, R.; Ebine, K.; Licht, J.D.; Munshi, H.G. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci. Rep. 2015, 5, 9489. [Google Scholar] [CrossRef]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; Neitzel, L.R.; Loganathan, S.N.; Tang, N.; Qin, L.; Crispi, E.E.; Guo, Y.; Knapp, S.; Beauchamp, R.D.; et al. The MAPK Pathway Regulates Intrinsic Resistance to BET Inhibitors in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2027–2037. [Google Scholar] [CrossRef]
- Ambrosini, G.; Do, C.; Tycko, B.; Realubit, R.B.; Karan, C.; Musi, E.; Carvajal, R.D.; Chua, V.; Aplin, A.E.; Schwartz, G.K. Inhibition of NF-kappaB-dependent signaling enhances sensitivity and overcomes resistance to BET inhibition in uveal melanoma. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Fulciniti, M.; Lin, C.Y.; Samur, M.K.; Lopez, M.A.; Singh, I.; Lawlor, M.A.; Szalat, R.E.; Ott, C.J.; Avet-Loiseau, H.; Anderson, K.C.; et al. Non-overlapping Control of Transcriptome by Promoter- and Super-Enhancer-Associated Dependencies in Multiple Myeloma. Cell Rep. 2018, 25, 3693–3705.e6. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nature Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Basu, T.N.; Gutmann, D.H.; Fletcher, J.A.; Glover, T.W.; Collins, F.S.; Downward, J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 1992, 356, 713–715. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef]
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D.R. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Sci. Rep. 2017, 7, 14992. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Haigentz, M., Jr.; Kim, M.; Sarta, C.; Lin, J.; Keresztes, R.S.; Culliney, B.; Gaba, A.G.; Smith, R.V.; Shapiro, G.I.; Chirieac, L.R.; et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol. 2012, 48, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.D.; Doroshow, J.H.; Wong, C.; et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: A California Cancer Consortium study. Clin. Cancer Res. 2008, 14, 7138–7142. [Google Scholar] [CrossRef] [PubMed]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharmacol. 2018, 81, 355–364. [Google Scholar] [CrossRef]
- Teknos, T.N.; Grecula, J.; Agrawal, A.; Old, M.O.; Ozer, E.; Carrau, R.; Kang, S.; Rocco, J.; Blakaj, D.; Diavolitsis, V.; et al. A phase 1 trial of Vorinostat in combination with concurrent chemoradiation therapy in the treatment of advanced staged head and neck squamous cell carcinoma. Invest. New Drugs 2018. [Google Scholar] [CrossRef]
- Pili, R.; Liu, G.; Chintala, S.; Verheul, H.; Rehman, S.; Attwood, K.; Lodge, M.A.; Wahl, R.; Martin, J.I.; Miles, K.M.; et al. Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in patients with clear-cell renal cell carcinoma: A multicentre, single-arm phase I/II clinical trial. Br. J. Cancer 2017, 116, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef]
- Schaukowitch, K.; Joo, J.Y.; Liu, X.; Watts, J.K.; Martinez, C.; Kim, T.K. Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 2014, 56, 29–42. [Google Scholar] [CrossRef]
- Liang, J.; Zhou, H.; Gerdt, C.; Tan, M.; Colson, T.; Kaye, K.M.; Kieff, E.; Zhao, B. Epstein–Barr virus super-enhancer eRNAs are essential for MYC oncogene expression and lymphoblast proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 14121. [Google Scholar] [CrossRef]
- Pnueli, L.; Rudnizky, S.; Yosefzon, Y.; Melamed, P. RNA transcribed from a distal enhancer is required for activating the chromatin at the promoter of the gonadotropin alpha-subunit gene. Proc. Natl. Acad. Sci. USA 2015, 112, 4369–4374. [Google Scholar] [CrossRef]
- Kim, Y.W.; Lee, S.; Yun, J.; Kim, A. Chromatin looping and eRNA transcription precede the transcriptional activation of gene in the β-globin locus. Biosci. Rep. 2015, 35, e00179. [Google Scholar] [CrossRef]
- Cheng, J.H.; Pan, D.Z.; Tsai, Z.T.; Tsai, H.K. Genome-wide analysis of enhancer RNA in gene regulation across 12 mouse tissues. Sci. Rep. 2015, 5, 12648. [Google Scholar] [CrossRef]
- McCleland, M.L.; Mesh, K.; Lorenzana, E.; Chopra, V.S.; Segal, E.; Watanabe, C.; Haley, B.; Mayba, O.; Yaylaoglu, M.; Gnad, F.; et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Investig. 2016, 126, 639–652. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, L.; Ren, S.; Wang, L.; Blackburn, P.R.; McNulty, M.S.; Gao, X.; Qiao, M.; Vessella, R.L.; Kohli, M.; et al. Activation of P-TEFb by Androgen Receptor-Regulated Enhancer RNAs in Castration-Resistant Prostate Cancer. Cell Rep. 2016, 15, 599–610. [Google Scholar] [CrossRef]
- Kaczkowski, B.; Tanaka, Y.; Kawaji, H.; Sandelin, A.; Andersson, R.; Itoh, M.; Lassmann, T.; Hayashizaki, Y.; Carninci, P.; Forrest, A.R. Transcriptome Analysis of Recurrently Deregulated Genes across Multiple Cancers Identifies New Pan-Cancer Biomarkers. Cancer Res. 2016, 76, 216–226. [Google Scholar] [CrossRef]
- Core, L.J.; Waterfall, J.J.; Lis, J.T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 2008, 322, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Schwalb, B.; Michel, M.; Zacher, B.; Fruhauf, K.; Demel, C.; Tresch, A.; Gagneur, J.; Cramer, P. TT-seq maps the human transient transcriptome. Science 2016, 352, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Mahat, D.B.; Kwak, H.; Booth, G.T.; Jonkers, I.H.; Danko, C.G.; Patel, R.K.; Waters, C.T.; Munson, K.; Core, L.J.; Lis, J.T. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc. 2016, 11, 1455–1476. [Google Scholar] [CrossRef]
- Chu, T.; Rice, E.J.; Booth, G.T.; Salamanca, H.H.; Wang, Z.; Core, L.J.; Longo, S.L.; Corona, R.J.; Chin, L.S.; Lis, J.T.; et al. Chromatin run-on and sequencing maps the transcriptional regulatory landscape of glioblastoma multiforme. Nat. Genet. 2018, 50, 1553–1564. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef]
- Parker, S.C.J.; Stitzel, M.L.; Taylor, D.L.; Orozco, J.M.; Erdos, M.R.; Akiyama, J.A.; van Bueren, K.L.; Chines, P.S.; Narisu, N.; Program, N.C.S.; et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. USA 2013, 110, 17921–17926. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.X.; Erdos, M.R.; Parker, S.C.J.; Collins, F.S. Motif signatures in stretch enhancers are enriched for disease-associated genetic variants. Epigenetics Chromatin 2015, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mathelier, A.; Zhang, X. Super-enhancers are transcriptionally more active and cell type-specific than stretch enhancers. Epigenetics 2018, 13, 910–922. [Google Scholar] [CrossRef]
- Hong, J.W.; Hendrix, D.A.; Levine, M.S. Shadow enhancers as a source of evolutionary novelty. Science 2008, 321, 1314. [Google Scholar] [CrossRef]
- Cao, K.; Collings, C.K.; Marshall, S.A.; Morgan, M.A.; Rendleman, E.J.; Wang, L.; Sze, C.C.; Sun, T.; Bartom, E.T.; Shilatifard, A. SET1A/COMPASS and shadow enhancers in the regulation of homeotic gene expression. Genes Dev. 2017, 31, 787–801. [Google Scholar] [CrossRef]
- Benabdallah, N.S.; Bickmore, W.A. Regulatory Domains and Their Mechanisms. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef]
- van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [PubMed]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.R.; Rubin, A.J.; Flynn, R.A.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef]
- Bretz, A.C.; Gittler, M.P.; Charles, J.P.; Gremke, N.; Eckhardt, I.; Mernberger, M.; Mandic, R.; Thomale, J.; Nist, A.; Wanzel, M.; et al. ΔNp63 activates the Fanconi anemia DNA repair pathway and limits the efficacy of cisplatin treatment in squamous cell carcinoma. Nucleic Acids Res. 2016, 44, 3204–3218. [Google Scholar] [CrossRef]
- Kumakura, Y.; Rokudai, S.; Iijima, M.; Altan, B.; Yoshida, T.; Bao, H.; Yokobori, T.; Sakai, M.; Sohda, M.; Miyazaki, T.; et al. Elevated expression of DeltaNp63 in advanced esophageal squamous cell carcinoma. Cancer Sci. 2017, 108, 2149–2155. [Google Scholar] [CrossRef]
- Coates, P.J.; Nenutil, R.; Holcakova, J.; Nekulova, M.; Podhorec, J.; Svoboda, M.; Vojtesek, B. p63 isoforms in triple-negative breast cancer: DeltaNp63 associates with the basal phenotype whereas TAp63 associates with androgen receptor, lack of BRCA mutation, PTEN and improved survival. Virchows Arch. 2018, 472, 351–359. [Google Scholar] [CrossRef]
- Orzol, P.; Nekulova, M.; Holcakova, J.; Muller, P.; Votesek, B.; Coates, P.J. DeltaNp63 regulates cell proliferation, differentiation, adhesion, and migration in the BL2 subtype of basal-like breast cancer. Tumour Biol. 2016, 37, 10133–10140. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdan, F.H.; Johnsen, S.A. Perturbing Enhancer Activity in Cancer Therapy. Cancers 2019, 11, 634. https://doi.org/10.3390/cancers11050634
Hamdan FH, Johnsen SA. Perturbing Enhancer Activity in Cancer Therapy. Cancers. 2019; 11(5):634. https://doi.org/10.3390/cancers11050634
Chicago/Turabian StyleHamdan, Feda H., and Steven A. Johnsen. 2019. "Perturbing Enhancer Activity in Cancer Therapy" Cancers 11, no. 5: 634. https://doi.org/10.3390/cancers11050634
APA StyleHamdan, F. H., & Johnsen, S. A. (2019). Perturbing Enhancer Activity in Cancer Therapy. Cancers, 11(5), 634. https://doi.org/10.3390/cancers11050634