Identification of a Clinically Relevant Signature for Early Progression in KRAS-Driven Lung Adenocarcinoma

 , ,
, , 
Abstract
:1. Introduction
2. Results
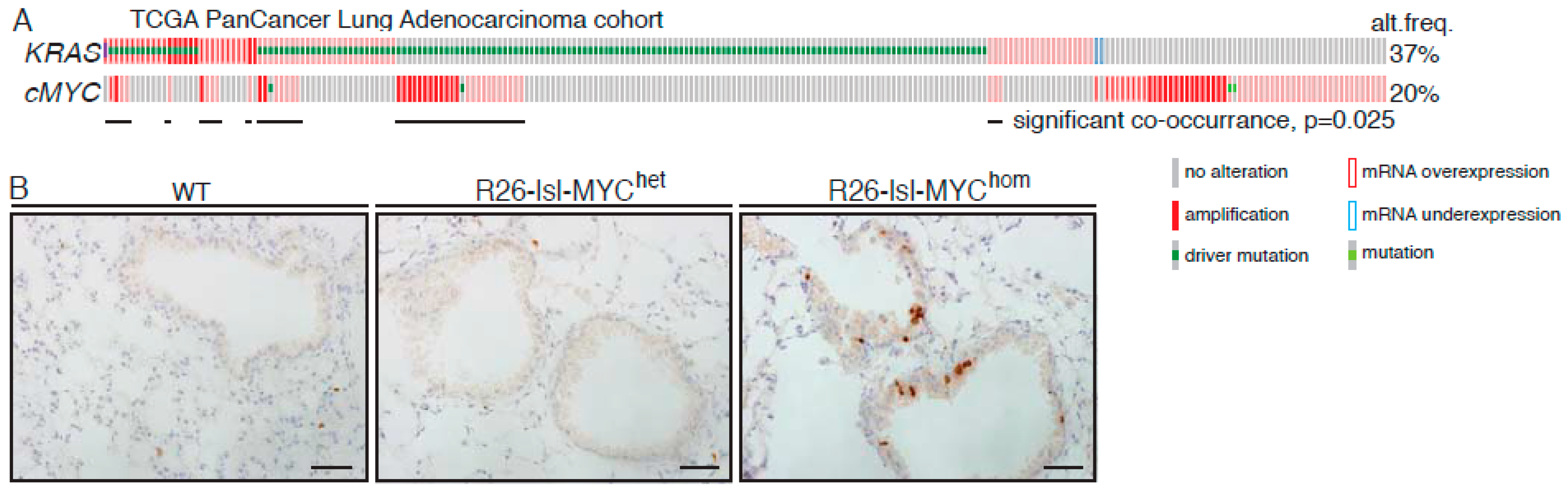
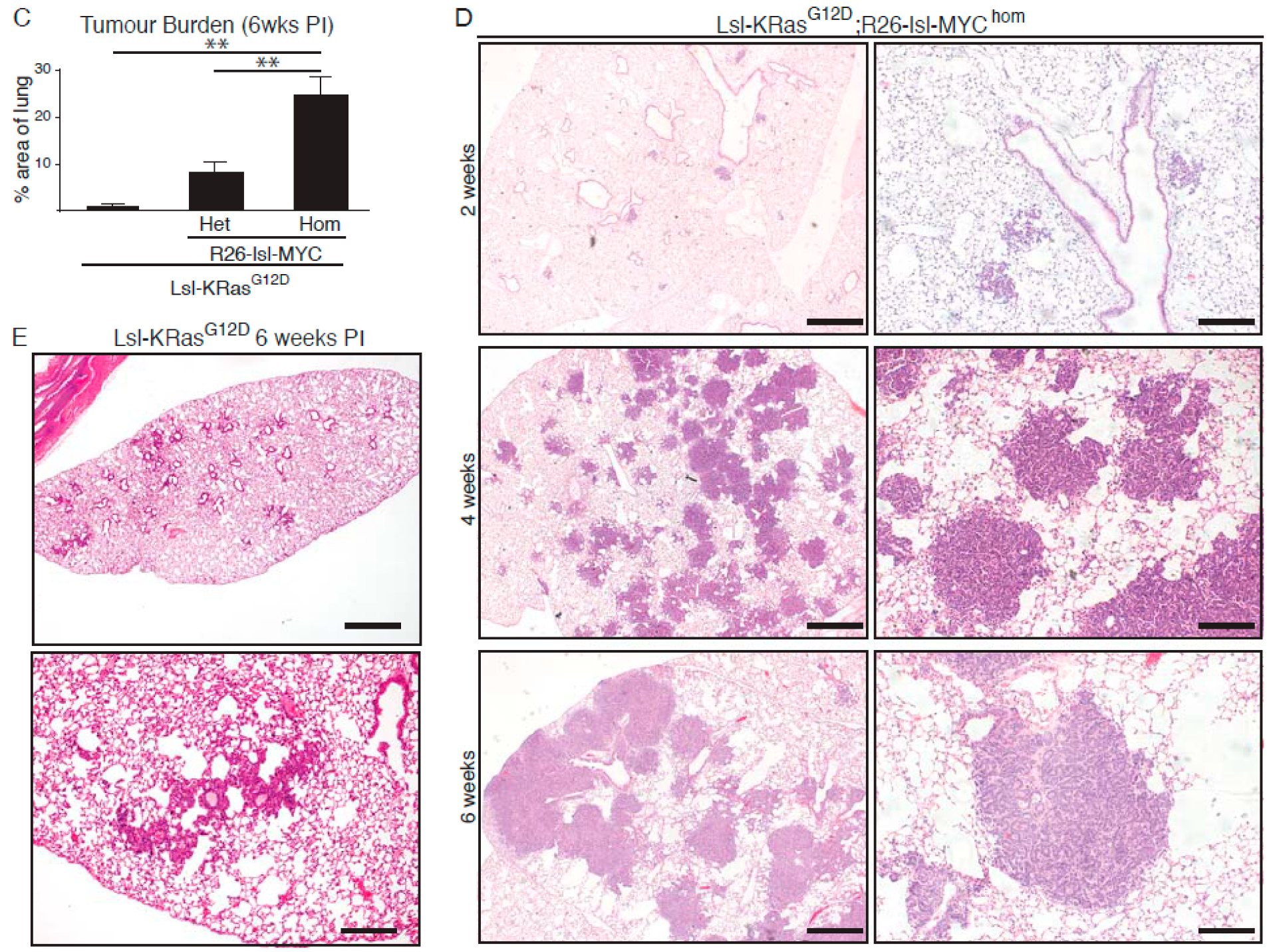
2.1. MYC Accelerates KRasG12D-Driven LuAd Development
2.2. A Transcriptomic Signature of KRAS LuAd Tumour Progression
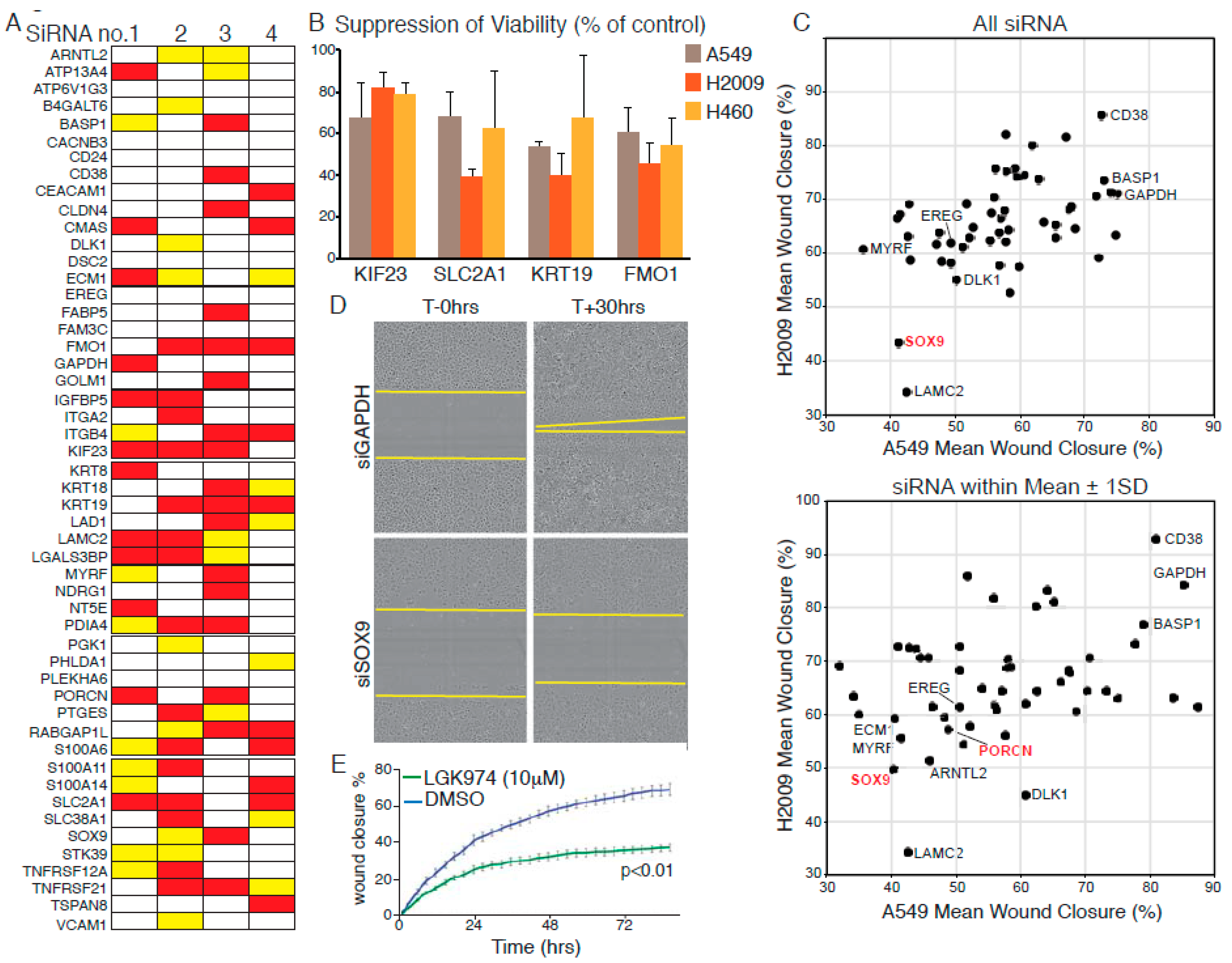
2.3. Functional Validation of p-Erk Associated Genes: Contribution to Cell Propagation
2.4. Functional Validation of p-Erk Associated Genes: WNT Signalling Contributes to Cell Motility
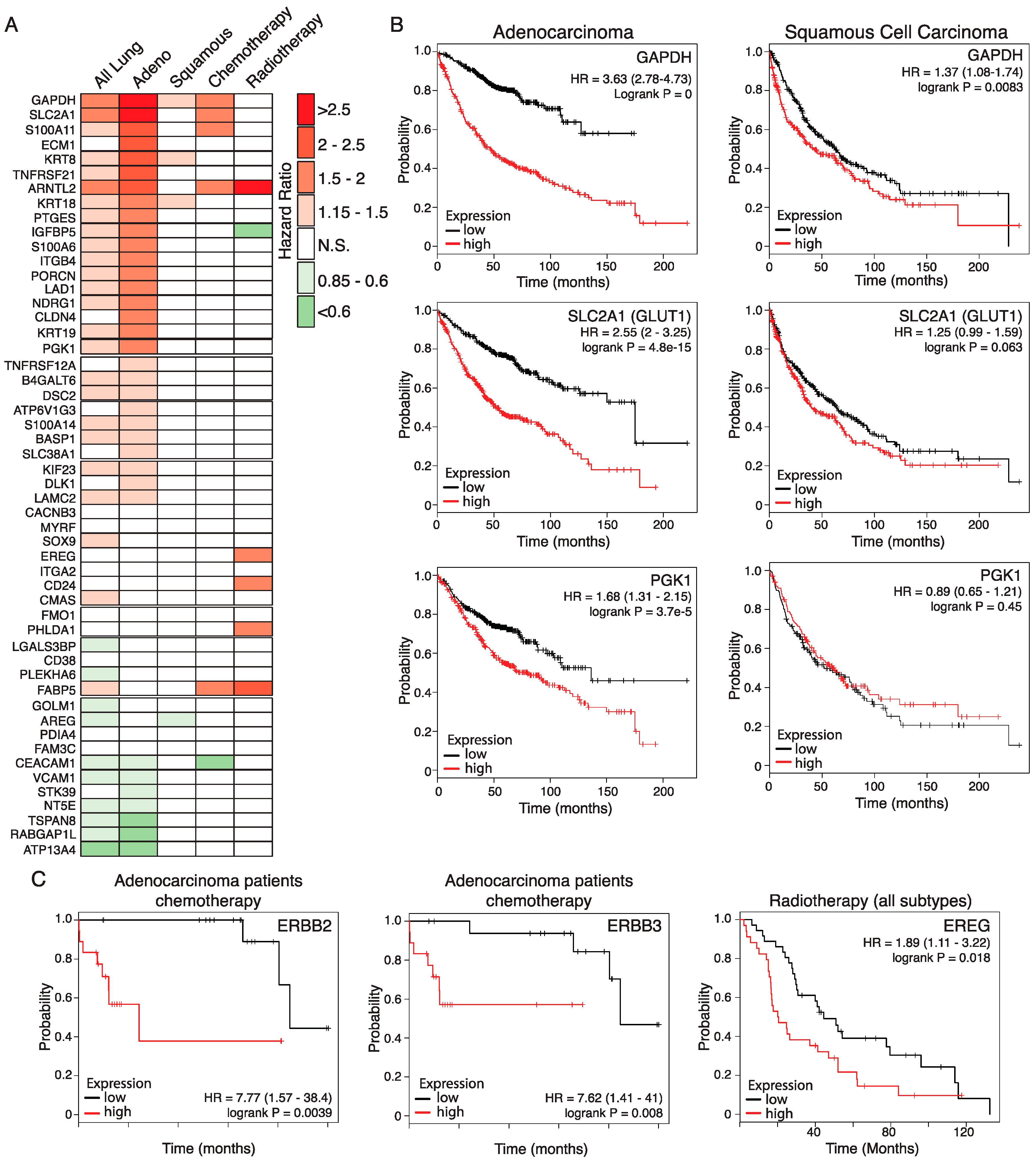
2.5. Relevance of the Murine Dataset for Human Pulmonary Adenocarcinoma
3. Discussion
4. Materials and Methods
4.1. Genetically Engineered Mice and Mouse Procedures
4.2. Immunohistochemistry and Tissue Analysis
4.3. Cell Culture and Related Assays
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kwon, M.C.; Berns, A. Mouse models for lung cancer. Mol. Oncol. 2013, 7, 165–177. [Google Scholar] [CrossRef]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Johnson, L. Using genetically engineered mouse models of cancer to aid drug development: An industry perspective. Clin. Cancer Res. 2006, 12, 5312–5328. [Google Scholar] [CrossRef]
- Singh, M.; Murriel, C.L.; Johnson, L. Genetically engineered mouse models: Closing the gap between preclinical data and trial outcomes. Cancer Res. 2012, 72, 2695–2700. [Google Scholar] [CrossRef]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumour initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Shaw, A.T.; Willis, N.A.; Silver, D.P.; Jackson, E.L.; Chang, S.; Mercer, K.L.; Grochow, R.; Hock, H.; Crowley, D.; et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 2004, 5, 375–387. [Google Scholar] [CrossRef]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumourigenesis. Nat. Cell Biol. 2007, 9, 493–505. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Jackson, E.L.; Olive, K.P.; Tuveson, D.A.; Bronson, R.; Crowley, D.; Brown, M.; Jacks, T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005, 65, 10280–10288. [Google Scholar] [CrossRef]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G.I. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002, 109, 321–334. [Google Scholar] [CrossRef]
- Dansen, T.B.; Whitfield, J.; Rostker, F.; Brown-Swigart, L.; Evan, G.I. Specific requirement for Bax, not Bak, in Myc-induced apoptosis and tumour suppression in vivo. J. Biol. Chem. 2006, 281, 10890–10895. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Lowe, S.W. Apoptosis and chemoresistance in transgenic cancer models. J. Mol. Med. (Berl) 2002, 80, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.; Prescott, J.; Shchors, K.; Hunt, A.; Soucek, L.; Dansen, T.B.; Swigart, L.B.; Evan, G.I. Bcl-xL gain of function and p19 ARF loss of function cooperate oncogenically with Myc in vivo by distinct mechanisms. Cancer Cell 2006, 10, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.J.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar] [CrossRef]
- Indra, A.K.; Warot, X.; Brocard, J.; Bornert, J.M.; Xiao, J.H.; Chambon, P.; Metzger, D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: Comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999, 27, 4324–4327. [Google Scholar] [CrossRef] [PubMed]
- Muthalagu, N.; Junttila, M.R.; Wiese, K.E.; Wolf, E.; Morton, J.; Bauer, B.; Evan, G.I.; Eilers, M.; Murphy, D.J. BIM is the primary mediator of MYC-induced apoptosis in multiple solid tissues. Cell Rep. 2014, 8, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.S.; Mason, S.M.; Athineos, D.; et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef]
- Sanchez-Aguilera, A.; Arranz, L.; Martin-Perez, D.; Garcia-Garcia, A.; Stavropoulou, V.; Kubovcakova, L.; Isern, J.; Martin-Salamanca, S.; Langa, X.; Skoda, R.C.; et al. Estrogen signalling selectively induces apoptosis of hematopoietic progenitors and myeloid neoplasms without harming steady-state hematopoiesis. Cell Stem Cell 2014, 15, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Neidler, S.; Murphy, D.J.; Institute of Cancer Sciences, University of Glasgow, Glasgo, UK. Personal communication, 2012.
- Nikitin, A.Y.; Alcaraz, A.; Anver, M.R.; Bronson, R.T.; Cardiff, R.D.; Dixon, D.; Fraire, A.E.; Gabrielson, E.W.; Gunning, W.T.; Haines, D.C.; et al. Classification of proliferative pulmonary lesions of the mouse: Recommendations of the mouse models of human cancers consortium. Cancer Res. 2004, 64, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Kruspig, B.; Monteverde, T.; Neidler, S.; Hock, A.; Kerr, E.; Nixon, C.; Clark, W.; Hedley, A.; Laing, S.; Coffelt, S.B.; et al. The ERBB network facilitates KRAS-driven lung tumourigenesis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Richards, W.G.; Staunton, J.; Li, C.; Monti, S.; Vasa, P.; Ladd, C.; Beheshti, J.; Bueno, R.; Gillette, M.; et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. USA 2001, 98, 13790–13795. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.E.; Troyanskaya, O.G.; Schluens, K.; Petersen, S.; Thaesler, Z.; Pacyna-Gengelbach, M.; van de Rijn, M.; Rosen, G.D.; Perou, C.M.; Whyte, R.I.; et al. Diversity of gene expression in adenocarcinoma of the lung. Proc. Natl. Acad. Sci. USA 2001, 98, 13784–13789. [Google Scholar] [CrossRef] [PubMed]
- Okayama, H.; Kohno, T.; Ishii, Y.; Shimada, Y.; Shiraishi, K.; Iwakawa, R.; Furuta, K.; Tsuta, K.; Shibata, T.; Yamamoto, S.; et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012, 72, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef]
- De Geus-Oei, L.F.; van der Heijden, H.F.; Corstens, F.H.; Oyen, W.J. Predictive and prognostic value of FDG-PET in nonsmall-cell lung cancer: A systematic review. Cancer 2007, 110, 1654–1664. [Google Scholar] [CrossRef]
- Moll, H.P.; Pranz, K.; Musteanu, M.; Grabner, B.; Hruschka, N.; Mohrherr, J.; Aigner, P.; Stiedl, P.; Brcic, L.; Laszlo, V.; et al. Afatinib restrains K-RAS-driven lung tumourigenesis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016, 531, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumourigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Port, J.; Muthalagu, N.; Raja, M.; Ceteci, F.; Monteverde, T.; Kruspig, B.; Hedley, A.; Kalna, G.; Lilla, S.; Neilson, L.; et al. Colorectal Tumours Require NUAK1 for Protection from Oxidative Stress. Cancer Discov. 2018, 8, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Hobor, S.; Bertotti, A.; Zecchin, D.; Huang, S.; Galimi, F.; Cottino, F.; Prahallad, A.; Grernrum, W.; Tzani, A.; et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014, 7, 86–93. [Google Scholar] [CrossRef]
- Mishra, R.; Hanker, A.B.; Garrett, J.T. Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget 2017, 8, 114371–114392. [Google Scholar] [CrossRef]
- Srinivas, S.; Watanabe, T.; Lin, C.S.; William, C.M.; Tanabe, Y.; Jessell, T.M.; Costantini, F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001, 1, 4. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Gene Name | Range p-ERK neg | Range p-ERK pos | Mean FC | FDR | Amp in NSCLC | OverEx in NSCLC |
|---|---|---|---|---|---|---|---|
| Ereg | Epiregulin | 5–39 | 227–376 | 24.82 | 2.78e-13 | Y | Y |
| Sox9 | SRY-like containing gene 9 | 6–31 | 39–330 | 15.79 | 7.48e-04 | Y | Y |
| Dlk1 | Delta-like 1 homolog | 63–400 | 341–2114 | 10.46 | 2.33e-26 | Y(SCC) | N |
| B4galt6 | UDP-Gal:betaGlcNAc beta 1,4-galactosyltransferase, 6 | 7–91 | 113–491 | 9.46 | 1.24e-03 | Y(SCC) | Y |
| Basp1 | Brain abundant, membrane attached signal protein 1 | 0–66 | 89–307 | 9.03 | 1.31e-04 | Y > 10% | Y |
| Itga2 | Integrin alpha 2 | 49–185 | 185–1239 | 6.79 | 2.47e-11 | <1% | Y |
| Cldn4 | Claudin 4 | 7–49 | 64–117 | 6.11 | 1.16e-02 | Y | Y |
| CD24a | CD24a antigen | 145–1040 | 1411–1580 | 5.94 | 1.4e-15 | No data | Y |
| Slc38A1 | Solute carrier family 38, member 1 | 60–153 | 248–716 | 5.68 | 1.17e-11 | Y | Y |
| Arntl2 | Aryl hydrocarbon receptor nuclear translocator-like 2 | 15–59 | 44–156 | 4.54 | 4.9e-02 | Y > 7% | Y |
| Dsc2 | Desmocollin | 14–148 | 62–302 | 4.43 | 4.51e-02 | Y(SCC) | Y |
| Areg | Amphiregulin | 156–234 | 526–831 | 4.25 | 4.98e-11 | Y | Y |
| S100a6 | S100 calcium binding protein A6 | 115–215 | 234–1017 | 4.00 | 1.51e-07 | Y > 14% | Y |
| Itgb4 | Integrin beta 4 | 31–89 | 81–310 | 3.92 | 2.29e-02 | Y | Y |
| Atp13a4 | ATPase type 13A4 | 242–399 | 711–1002 | 3.92 | 3.03e-14 | Y > 25% | Y |
| Kif23 | Kinesin family member 23 | 151–359 | 313–994 | 3.76 | 8.81e-07 | <1% | Y |
| Porcn | Porcupine homolog | 38–52 | 76–182 | 3.59 | 4.85e-03 | <1% | N |
| Krt8 | Keratin 8 | 67–380 | 489–699 | 3.57 | 1.87e-05 | Y | Y |
| Vcam1 | Vascular cell adhesion molecule 1 | 11–133 | 75–442 | 3.57 | 3.59e-02 | <1% | N |
| Krt18 | Keratin 18 | 503–827 | 1192–1850 | 3.50 | 1.8e-11 | Y | Y |
| Lamc2 | Laminin gamma 2 | 457–677 | 1104–1825 | 3.48 | 2.56e-09 | Y > 7% | Y |
| Tnfrsf12a | Tumour necrosis factor receptor superfamily, 12a | 30–54 | 86–135 | 3.42 | 1.47e-02 | Y | Y |
| Tspan8 | Tetraspanin 8 | 329–570 | 635–1848 | 3.41 | 5.23e-06 | Y > 5% | Y |
| Atp6v1g3 | ATPase H+ transporting, lysosomal V1 subunit g3 | 46–94 | 91–232 | 3.40 | 2.8e-03 | Y > 7% | N |
| Ecm1 | Extracellular matrix protein 1 | 44–79 | 100–173 | 3.25 | 7.13e-03 | Y > 16% | Y |
| S100a14 | S100 calcium binding protein A14 | 89–210 | 218-530 | 3.18 | 6.52e-04 | Y > 14% | Y |
| Lgals3bp | Lectin, galactoside-binding, soluble, 3 binding protein | 178–333 | 472–679 | 3.16 | 2.09e-05 | Y | Y |
| Nt5e | 5’ nucleotidase, ecto | 148–339 | 462–697 | 3.04 | 1.2e-05 | Y(SCC) | Y |
| Lad1 | Ladinin 1 | 141–344 | 401–619 | 3.01 | 2.92e-05 | Y > 7% | Y |
| Igfbp5 | Insulin-like growth factor binding protein 5 | 296–422 | 414–1197 | 2.99 | 1.36e-05 | <1% | Y |
| Pdia4 | Protein disulphide isomerase associated 4 | 966–1887 | 2519–3183 | 2.98 | 3.16e-05 | Y | Y |
| CD38 | CD38 antigen | 37–64 | 77–167 | 2.97 | 3.82e-02 | <1% | Y |
| Krt19 | Keratin 19 | 203–469 | 664–969 | 2.96 | 2.06e-06 | <1% | Y |
| Slc2a1 | Solute carrier family 2, member 1 | 57–114 | 125–249 | 2.95 | 1.11e-02 | Y | Y |
| Myrf | Myelin regulatory factor | 57–163 | 95–474 | 2.92 | 1.26e-02 | <1% | Y |
| Plekha6 | Pleckstrin homology domain containing, family A, 6 | 185–353 | 490–692 | 2.91 | 3.34e-05 | <1% | Y |
| Ndrg1 | N-MYC downstream regulated gene 1 | 77–333 | 208–716 | 2.84 | 7.71e-03 | Y > 5% | Y |
| Rabgap1l | RAB GTPase activating protein 1-like | 1211–1754 | 2578–4099 | 2.83 | 3.95e-05 | Y > 8% | N |
| Phlda1 | Pleckstrin homology-like domain family A, member 1 | 105–226 | 175–402 | 2.82 | 7.01e-04 | <1% | Y |
| Gapdh | Glyceraldehyde-3-phosphate dehydrogenase | 79–106 | 136–216 | 2.80 | 1.08e-02 | Y(SCC) | Y |
| Fabp5 | Fatty acid binding protein 5, epidermal | 78-175 | 115–410 | 2.79 | 6.15e-03 | Y | Y |
| Cmas | Cytidine monophospho-N-acetylneuraminic acid synthetase | 49–106 | 108–283 | 2.75 | 4.21e-02 | Y > 5% | Y |
| Golm1 | Golgi membrane protein 1 | 509–1420 | 1343–2171 | 2.75 | 3.84e-05 | N | Y |
| Ptges | Prostaglandin E synthase | 158–286 | 158–286 | 2.73 | 5.26e-03 | Y | Y |
| Stk39 | Serine/threonine kinase 39 | 304–659 | 872–1127 | 2.71 | 2.21e-06 | Y | Y |
| Fam3c | Family with sequence similarity 3, member C | 351–642 | 673–1078 | 2.65 | 6.4e-09 | Y | N |
| Pgk1 | Phosphoglycerate kinase 1 | 138–178 | 220–425 | 2.61 | 1.23e-03 | <1% | Y |
| Cacnb3 | Calcium channel, voltage dependent, beta 3 | 56–87 | 78–147 | 2.59 | 3.7e-02 | Y | N |
| Tnfrsf21 | Tumour necrosis factor receptor superfamily, 21 | 121–238 | 232–348 | 2.57 | 1.58e-03 | Y | Y |
| Fmo1 | Flavin containing monooxygenase 1 | 174–256 | 186–1098 | 2.57 | 7.51e-03 | Y > 7% | Y |
| S100a11 | S100 calcium binding protein A11 | 245–696 | 594–1252 | 2.53 | 1.21e-04 | Y > 15% | Y |
| Ceacam1 | Carcinoembryonic antigen-related cell adhesion molecule 1 | 326–608 | 648–1286 | 2.50 | 1.08e-05 | Y | Y |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neidler, S.; Kruspig, B.; Hewit, K.; Monteverde, T.; Gyuraszova, K.; Braun, A.; Clark, W.; James, D.; Hedley, A.; Nieswandt, B.; et al. Identification of a Clinically Relevant Signature for Early Progression in KRAS-Driven Lung Adenocarcinoma. Cancers 2019, 11, 600. https://doi.org/10.3390/cancers11050600
Neidler S, Kruspig B, Hewit K, Monteverde T, Gyuraszova K, Braun A, Clark W, James D, Hedley A, Nieswandt B, et al. Identification of a Clinically Relevant Signature for Early Progression in KRAS-Driven Lung Adenocarcinoma. Cancers. 2019; 11(5):600. https://doi.org/10.3390/cancers11050600
Chicago/Turabian StyleNeidler, Sarah, Björn Kruspig, Kay Hewit, Tiziana Monteverde, Katarina Gyuraszova, Attila Braun, William Clark, Daniel James, Ann Hedley, Bernhard Nieswandt, and et al. 2019. "Identification of a Clinically Relevant Signature for Early Progression in KRAS-Driven Lung Adenocarcinoma" Cancers 11, no. 5: 600. https://doi.org/10.3390/cancers11050600
APA StyleNeidler, S., Kruspig, B., Hewit, K., Monteverde, T., Gyuraszova, K., Braun, A., Clark, W., James, D., Hedley, A., Nieswandt, B., Shanks, E., Dick, C., & Murphy, D. J. (2019). Identification of a Clinically Relevant Signature for Early Progression in KRAS-Driven Lung Adenocarcinoma. Cancers, 11(5), 600. https://doi.org/10.3390/cancers11050600