Integrative In Vivo Drug Testing Using Gene Expression Signature and Patient-Derived Xenografts from Treatment-Refractory HER2 Positive and Triple-Negative Subtypes of Breast Cancer


 and
and
Abstract
1. Introduction
2. Results
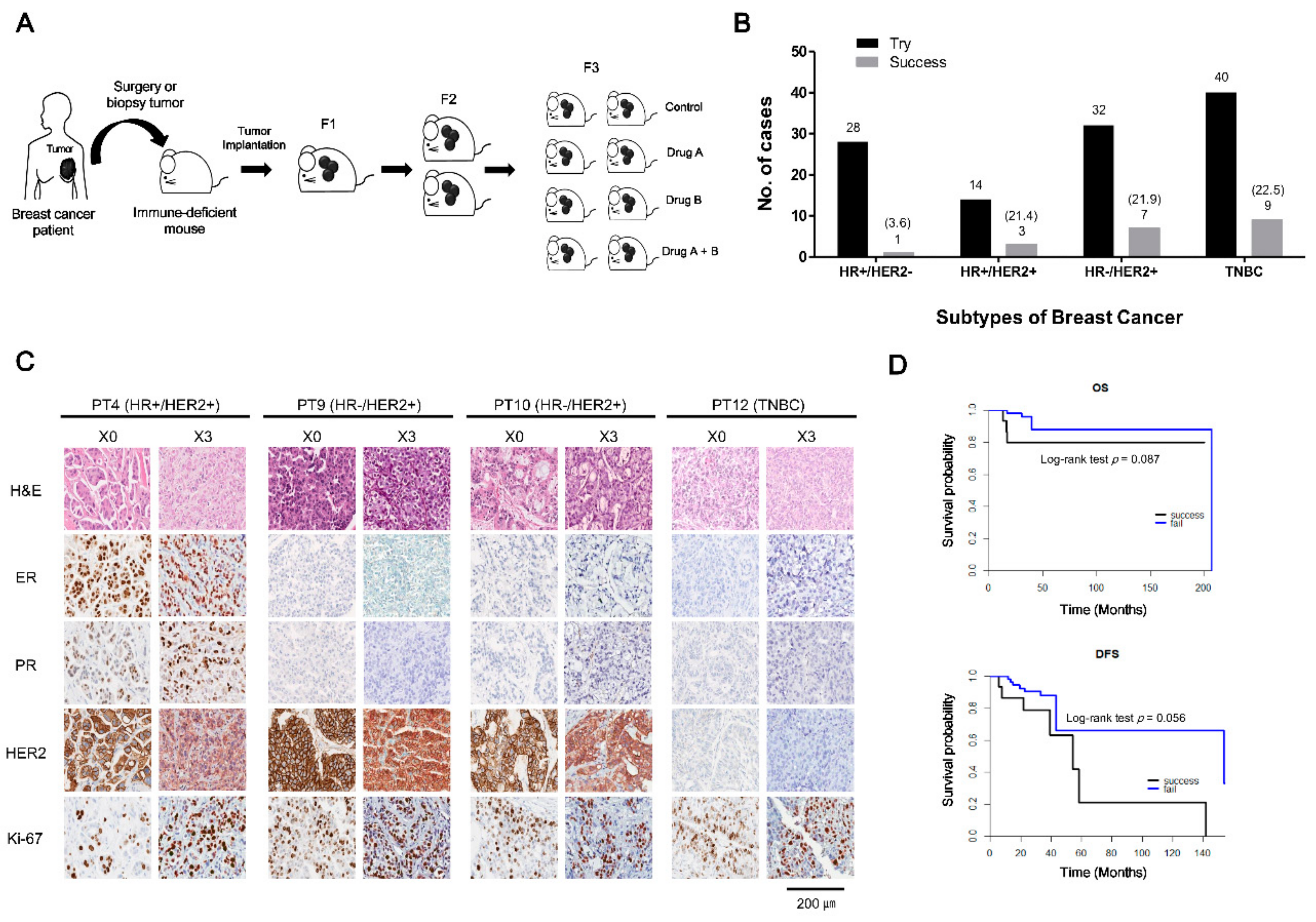
2.1. Patient Characteristics and PDX Establishment
2.2. Histologic Characteristics of Patient Breast Cancers and Their Corresponding Xenografts
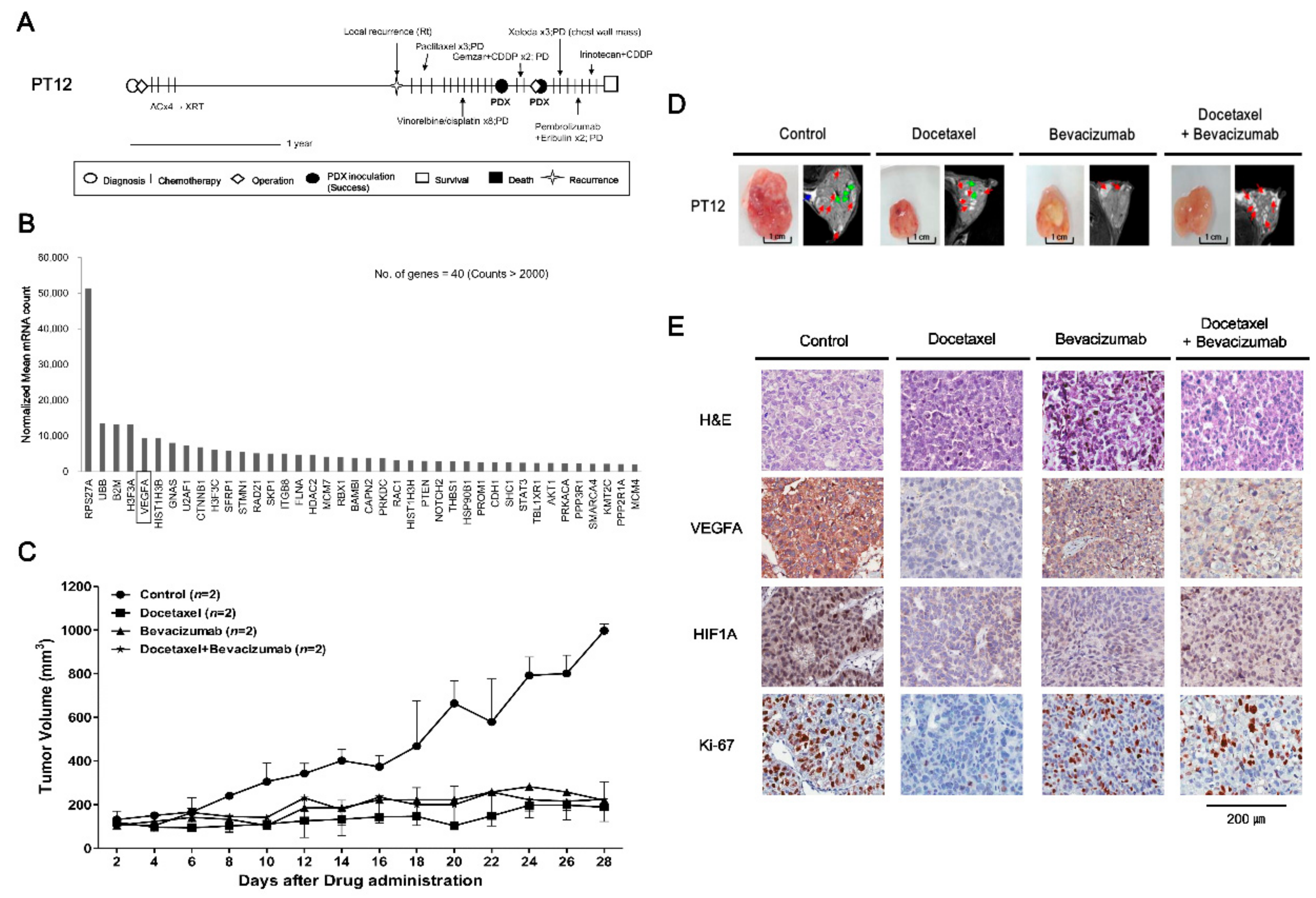
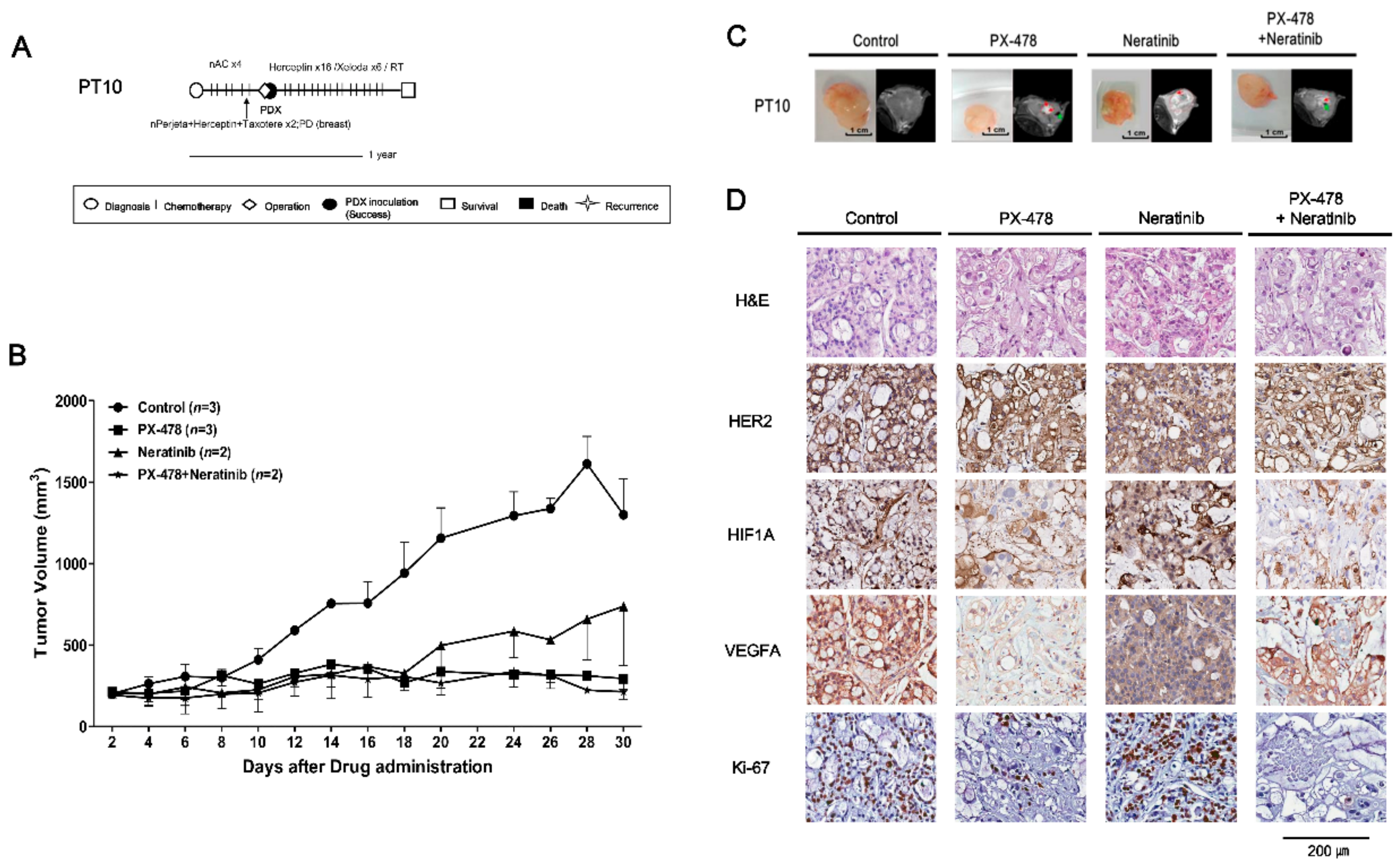
2.3. Drugs Response in PDX Models: TNBC Cases
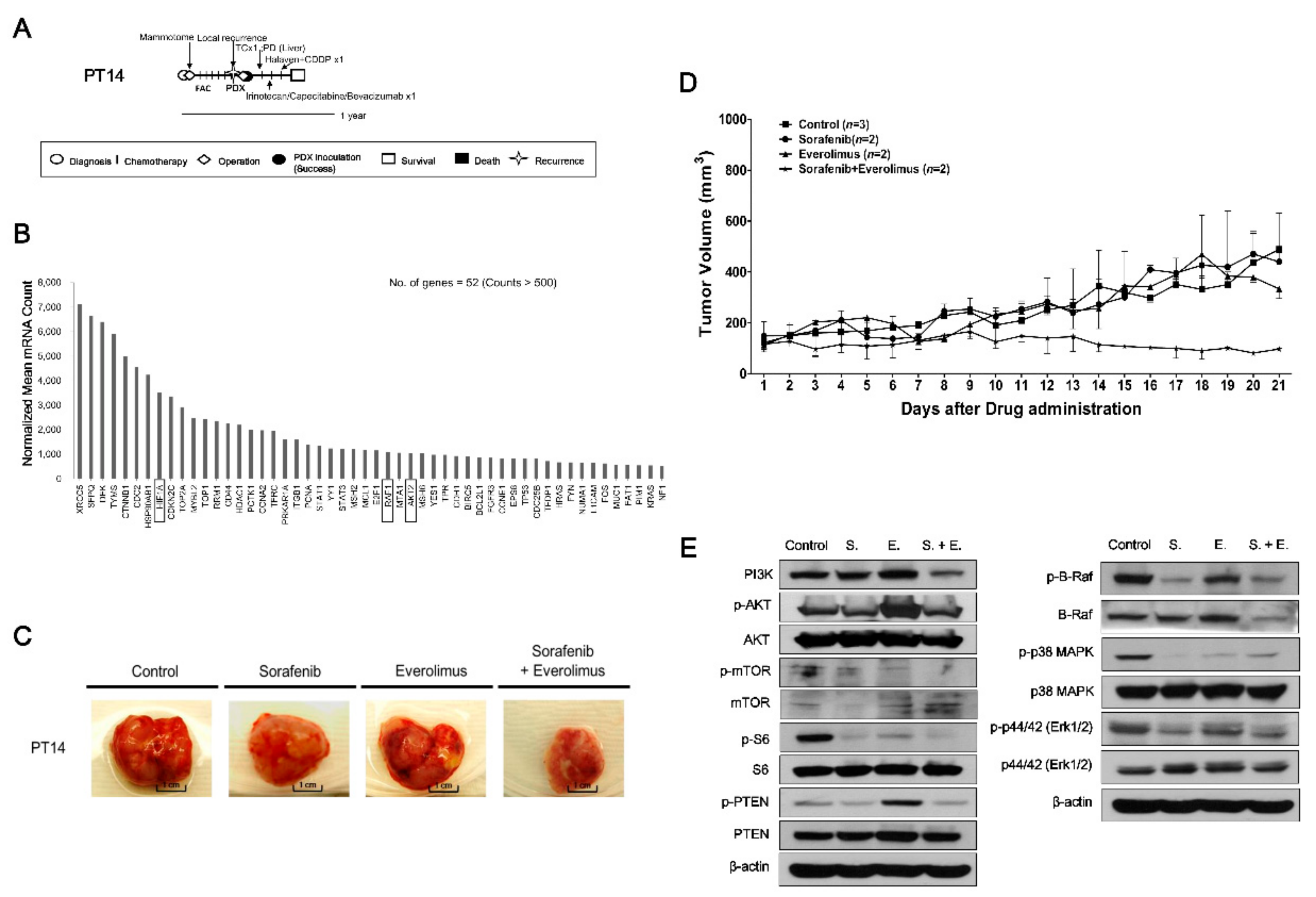
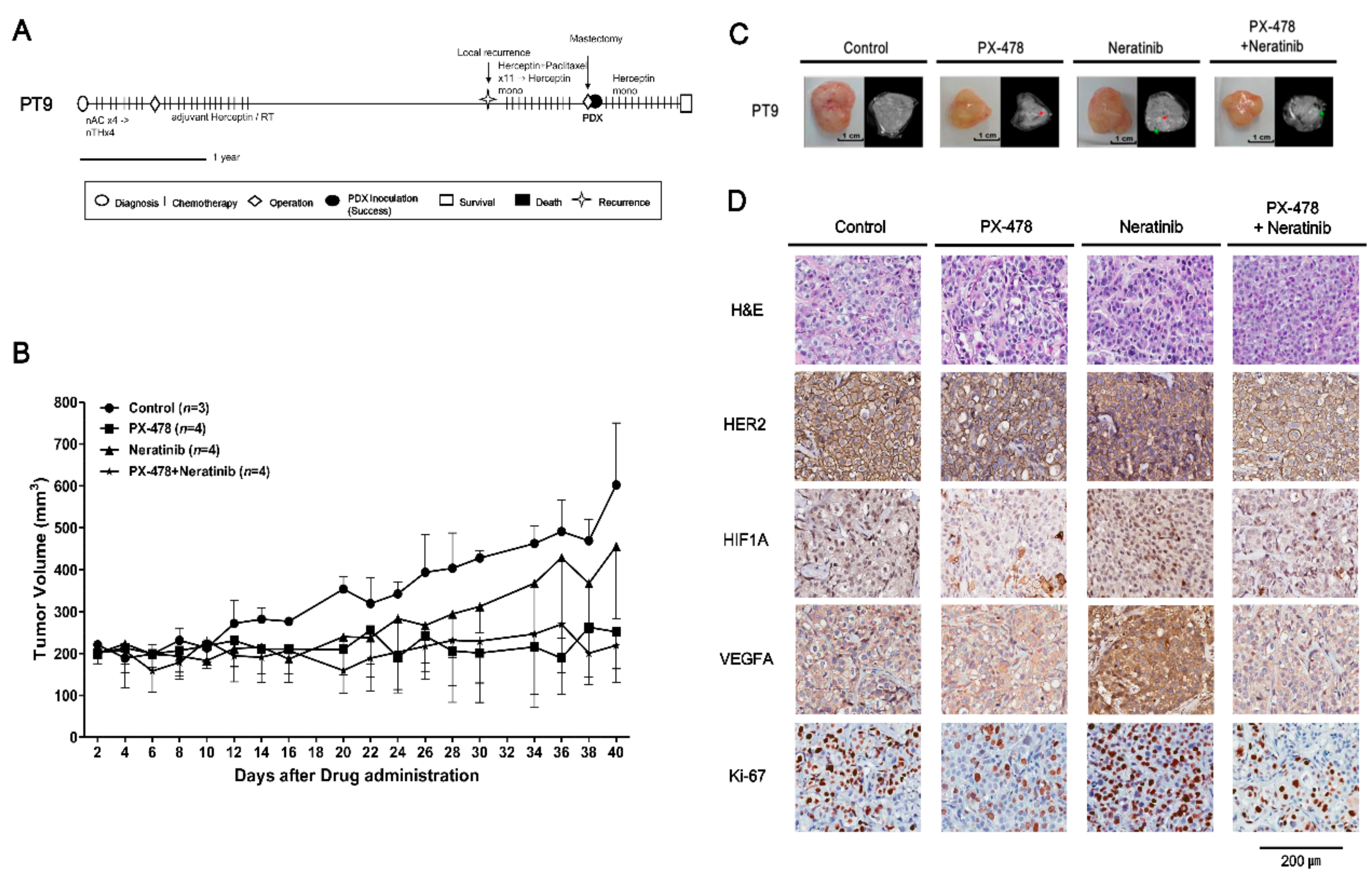
2.4. Evaluating Anticancer Drug Effects in Trastuzumab-Exposed PDX Models: Cases of HR−/HER2+ Breast Cancer
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Generation of PDX Breast Cancer Models
4.3. In Vivo Drug Treatment
4.4. Histologic Evaluation of PDX Tumors
4.5. Detection of Target Genes in PDX Tumors Using Nanostring multigene panels
4.6. Evaluation of Target Protein Responses to Drug Treatment by Western Blotting
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- May, M. Cancer research with a human touch. Nature 2018, 556, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Seol, H.S.; Chang, S. The Generation and Application of Patient-Derived Xenograft Model for Cancer Research. Cancer Res. Treat. 2018, 50, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Whittle, J.R.; Lewis, M.T.; Lindeman, G.J.; Visvader, J.E. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015, 17, 17. [Google Scholar] [CrossRef]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef] [PubMed]
- Eyre, R.; Alferez, D.G.; Spence, K.; Kamal, M.; Shaw, F.L.; Simoes, B.M.; Santiago-Gomez, A.; Sarmiento-Castro, A.; Bramley, M.; Absar, M.; et al. Patient-derived Mammosphere and Xenograft Tumour Initiation Correlates with Progression to Metastasis. J. Mammary Gland Biol. Neoplasia 2016, 21, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Kabos, P.; Finlay-Schultz, J.; Li, C.; Kline, E.; Finlayson, C.; Wisell, J.; Manuel, C.A.; Edgerton, S.M.; Harrell, J.C.; Elias, A.; et al. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res. Treat 2012, 135, 415–432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cohen, A.L.; Krishnakumar, S.; Wapnir, I.L.; Veeriah, S.; Deng, G.; Coram, M.A.; Piskun, C.M.; Longacre, T.A.; Herrler, M.; et al. Patient-derived xenografts of triple-negative breast cancer reproduce molecular features of patient tumors and respond to mTOR inhibition. Breast Cancer Res. 2014, 16, R36. [Google Scholar] [CrossRef]
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Reyal, F.; Guyader, C.; Decraene, C.; Lucchesi, C.; Auger, N.; Assayag, F.; De Plater, L.; Gentien, D.; Poupon, M.F.; Cottu, P.; et al. Molecular profiling of patient-derived breast cancer xenografts. Breast Cancer Res. 2012, 14, R11. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Rowe, A.; Senthebane, D.A.; AlMazyadi, M.A.M.; Patten, V.; Parker, M.I. Three-Dimensional Organoids in Cancer Research: The Search for the Holy Grail of Preclinical Cancer Modeling. OMICS 2018, 22, 733–748. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Niu, G.; Chen, X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr. Drug Targets 2010, 11, 1000–1017. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Montero, A.J.; Escobar, M.; Lopes, G.; Gluck, S.; Vogel, C. Bevacizumab in the treatment of metastatic breast cancer: Friend or foe? Curr. Oncol. Rep. 2012, 14, 1–11. [Google Scholar] [CrossRef]
- Tiainen, L.; Tanner, M.; Lahdenpera, O.; Vihinen, P.; Jukkola, A.; Karihtala, P.; Paunu, N.; Huttunen, T.; Kellokumpu-Lehtinen, P.L. Bevacizumab Combined with Docetaxel or Paclitaxel as First-line Treatment of HER2-negative Metastatic Breast Cancer. Anticancer Res. 2016, 36, 6431–6438. [Google Scholar] [CrossRef] [PubMed]
- Pories, S.E.; Wulf, G.M. Evidence for the role of bevacizumab in the treatment of advanced metastatic breast cancer: A review. Breast Cancer (Dove Med. Press) 2010, 2, 37–44. [Google Scholar] [CrossRef]
- Koh, M.Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R.R.; Kirkpatrick, D.L.; Powis, G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2008, 7, 90–100. [Google Scholar] [CrossRef]
- Macpherson, G.R.; Figg, W.D. Small molecule-mediated anti-cancer therapy via hypoxia-inducible factor-1 blockade. Cancer Biol. Ther. 2004, 3, 503–504. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.; Williams, R.; Kirkpatrick, L.; Paine-Murrieta, G.; Powis, G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2004, 3, 233–244. [Google Scholar]
- Choi, S.Y.; Lin, D.; Gout, P.W.; Collins, C.C.; Xu, Y.; Wang, Y. Lessons from patient-derived xenografts for better in vitro modeling of human cancer. Adv. Drug Deliv. Rev. 2014, 79, 222–237. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Foster, B.A.; Young, J.; Takabe, K. Current Update of Patient-Derived Xenograft Model for Translational Breast Cancer Research. J. Mammary Gland Biol. Neoplasia 2017, 22, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, E.; Vincent-Salomon, A.; Auger, N.; Degeorges, A.; Assayag, F.; de Cremoux, P.; de Plater, L.; Guyader, C.; De Pinieux, G.; Judde, J.G.; et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin. Cancer Res. 2007, 13, 3989–3998. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.G.; Oh, K.; Lee, J.; Lee, M.; Kim, J.Y.; Yoo, T.K.; Seo, M.W.; Park, A.K.; Ryu, H.S.; Jung, E.J.; et al. Prognostic and functional importance of the engraftment-associated genes in the patient-derived xenograft models of triple-negative breast cancers. Breast Cancer Res. Treat 2015, 154, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Qin, B.; Moyer, A.M.; Sinnwell, J.P.; Thompson, K.J.; Copland, J.A., 3rd; Marlow, L.A.; Miller, J.L.; Yin, P.; Gao, B.; et al. Establishing and characterizing patient-derived xenografts using pre-chemotherapy percutaneous biopsy and post-chemotherapy surgical samples from a prospective neoadjuvant breast cancer study. Breast Cancer Res. 2017, 19, 130. [Google Scholar] [CrossRef]
- Kuperwasser, C.; Chavarria, T.; Wu, M.; Magrane, G.; Gray, J.W.; Carey, L.; Richardson, A.; Weinberg, R.A. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 4966–4971. [Google Scholar] [CrossRef]
- DeRose, Y.S.; Gligorich, K.M.; Wang, G.; Georgelas, A.; Bowman, P.; Courdy, S.J.; Welm, A.L.; Welm, B.E. Patient-derived models of human breast cancer: Protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr. Protoc. Pharm. 2013, 60, 14–23. [Google Scholar] [CrossRef]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J. Everolimus. Clin. Cancer Res. 2010, 16, 1368–1372. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef] [PubMed]
- Whelan, K.A.; Schwab, L.P.; Karakashev, S.V.; Franchetti, L.; Johannes, G.J.; Seagroves, T.N.; Reginato, M.J. The oncogene HER2/neu (ERBB2) requires the hypoxia-inducible factor HIF-1 for mammary tumor growth and anoikis resistance. J. Biol. Chem. 2013, 288, 15865–15877. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Imanaka, N.; Chen, J.; Griffin, J.D. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br. J. Cancer 2010, 102, 351–360. [Google Scholar] [CrossRef]
- West, M.; Blanchette, C.; Dressman, H.; Huang, E.; Ishida, S.; Spang, R.; Zuzan, H.; Olson, J.A.; Marks, J.R.; Nevins, J.R. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc. Natl. Acad. Sci. USA 2001, 98, 11462–11467. [Google Scholar] [CrossRef]
- Jwa, E.; Shin, K.H.; Kim, J.Y.; Park, Y.H.; Jung, S.Y.; Lee, E.S.; Park, I.H.; Lee, K.S.; Ro, J.; Kim, Y.J.; et al. Locoregional Recurrence by Tumor Biology in Breast Cancer Patients after Preoperative Chemotherapy and Breast Conservation Treatment. Cancer Res. Treat 2016, 48, 1363–1372. [Google Scholar] [CrossRef]
- Lee, M.H.; Jung, S.Y.; Kang, S.H.; Song, E.J.; Park, I.H.; Kong, S.Y.; Kwon, Y.M.; Lee, K.S.; Kang, H.S.; Lee, E.S. The Significance of Serum HER2 Levels at Diagnosis on Intrinsic Subtype-Specific Outcome of Operable Breast Cancer Patients. PLoS ONE 2016, 11, e0163370. [Google Scholar] [CrossRef]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Total (%) | Engrafted (%) | Failed (%) | p-Value |
|---|---|---|---|---|
| Number of patients | 114 (100) | 20 (17.5) | 94 (82.5) | |
| Age at collection (years) | 49.60 ± 11.60 | 46.40 ± 13.69 | 49.94 ± 10.90 | 0.175 (b) |
| Tissue source | ||||
| Surgery | 57 (50.0) | 14 (24.6) | 43 (75.4) | 0.085 (c) |
| Biopsy | 57 (50.0) | 6 (10.5) | 51 (89.5) | |
| Stage | ||||
| I | 9 (7.9) | 1 (11.1) | 8 (88.9) | 0.039 (d) |
| II | 47 (41.2) | 5 (10.6) | 42(89.4) | |
| III | 48 (42.1) | 9 (18.7) | 39(81.3) | |
| IV | 10 (8.8) | 5 (50.0) | 5 (50.0) | |
| Chemotherapy resistance (a) | ||||
| Yes | 28 (24.6) | 13 (46.4) | 15 (53.6) | <0.001 (d) |
| No | 86 (74.6) | 7 (8.1) | 79 (91.9) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, J.-S.; Sim, S.H.; Park, I.H.; Lee, E.G.; Lee, E.S.; Kim, Y.-H.; Kwon, Y.; Kong, S.-Y.; Lee, K.S. Integrative In Vivo Drug Testing Using Gene Expression Signature and Patient-Derived Xenografts from Treatment-Refractory HER2 Positive and Triple-Negative Subtypes of Breast Cancer. Cancers 2019, 11, 574. https://doi.org/10.3390/cancers11040574
Ryu J-S, Sim SH, Park IH, Lee EG, Lee ES, Kim Y-H, Kwon Y, Kong S-Y, Lee KS. Integrative In Vivo Drug Testing Using Gene Expression Signature and Patient-Derived Xenografts from Treatment-Refractory HER2 Positive and Triple-Negative Subtypes of Breast Cancer. Cancers. 2019; 11(4):574. https://doi.org/10.3390/cancers11040574
Chicago/Turabian StyleRyu, Jin-Sun, Sung Hoon Sim, In Hae Park, Eun Gyeong Lee, Eun Sook Lee, Yun-Hee Kim, Youngmee Kwon, Sun-Young Kong, and Keun Seok Lee. 2019. "Integrative In Vivo Drug Testing Using Gene Expression Signature and Patient-Derived Xenografts from Treatment-Refractory HER2 Positive and Triple-Negative Subtypes of Breast Cancer" Cancers 11, no. 4: 574. https://doi.org/10.3390/cancers11040574
APA StyleRyu, J.-S., Sim, S. H., Park, I. H., Lee, E. G., Lee, E. S., Kim, Y.-H., Kwon, Y., Kong, S.-Y., & Lee, K. S. (2019). Integrative In Vivo Drug Testing Using Gene Expression Signature and Patient-Derived Xenografts from Treatment-Refractory HER2 Positive and Triple-Negative Subtypes of Breast Cancer. Cancers, 11(4), 574. https://doi.org/10.3390/cancers11040574