Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy



Abstract
:1. Isocitrate Dehydrogenase Enzymes
2. IDHs Genetic Alterations in Cancer
3. IDHs Genetic Alterations in Genetic Diseases
4. Downregulation of Wild-Type IDH1 in Cancers
5. Overexpression of Wild-Type IDH1 in Cancers
6. Downregulation of Wild-Type IDH2 in Cancers
7. Overexpression of Wild-Type IDH2 in Cancers
8. Wild-Type IDH3 Downregulation/Overexpression in Cancer
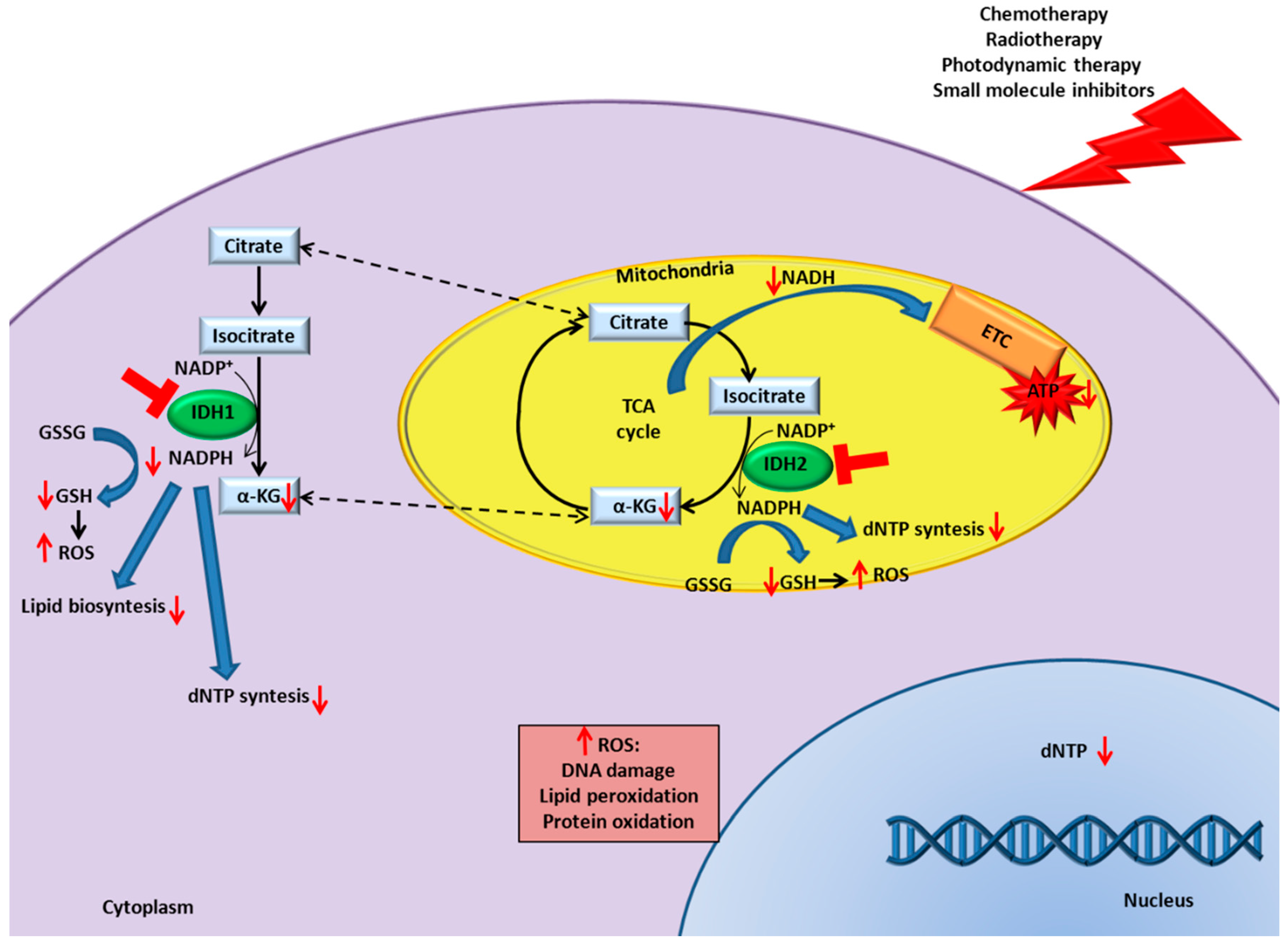
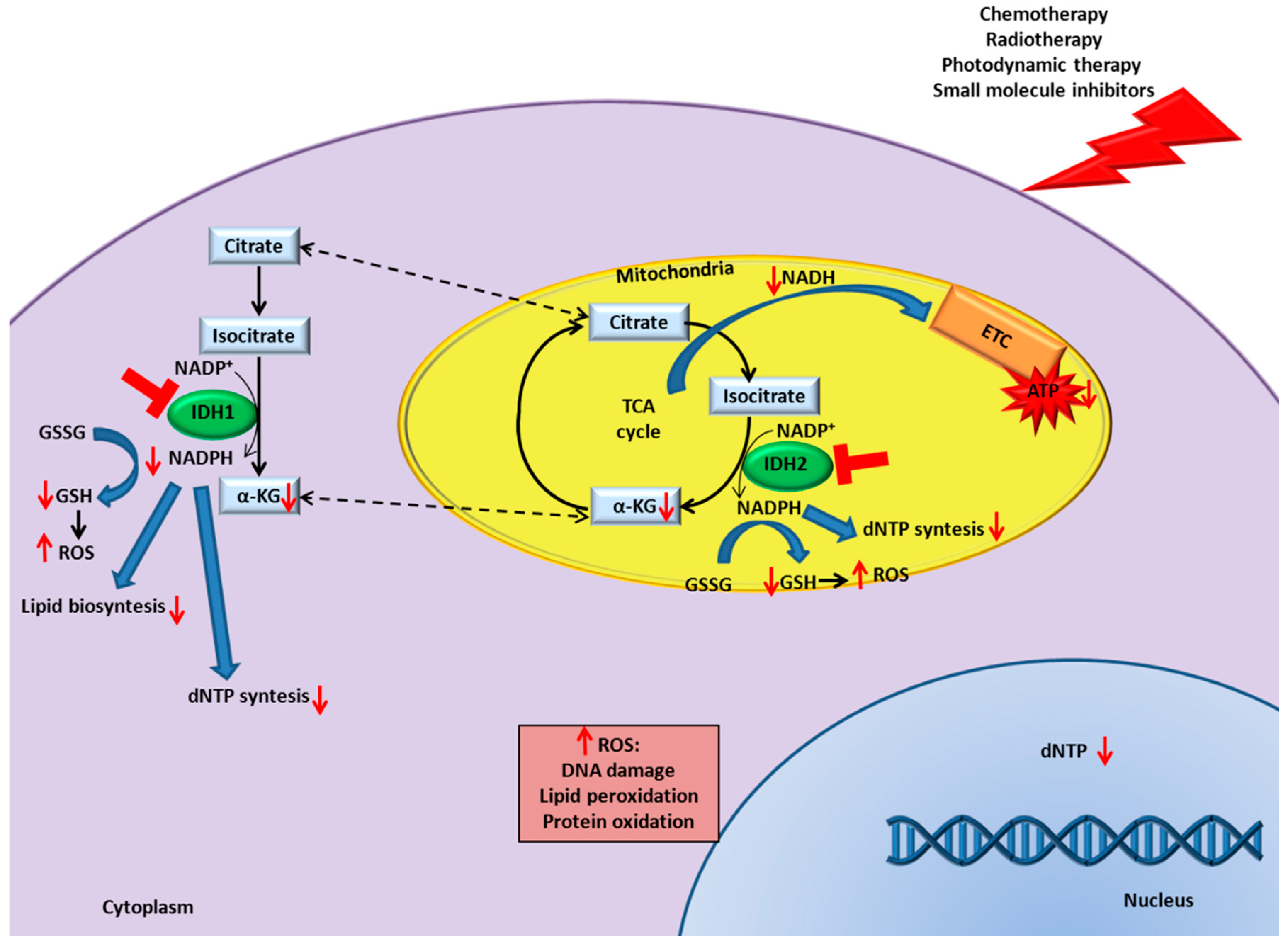
9. IDH Inhibition in Cancer Enhances Responsiveness to Canonical Therapies
9.1. Chemotherapy
9.2. Radiotherapy
9.3. Photodynamic Therapy
9.4. Small Molecule Inhibitors
10. Concluding Remarks and Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Geisbrecht, B.V.; Gould, S.J. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J. Biol. Chem. 1999, 274, 30527–30533. [Google Scholar] [CrossRef]
- Ma, T.; Peng, Y.; Huang, W.; Liu, Y.; Ding, J. The β and γ subunits play distinct functional roles in the α2βγ heterotetramer of human NAD-dependent isocitrate dehydrogenase. Sci. Rep. 2017, 7, 41882. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the Warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef]
- Fendt, S.-M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.R.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Heiden, M.G. Vander; Stephanopoulos, G. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 2236. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. NAD+/NADH and NADP+/NADPH in Cellular Functions and Cell Death: Regulation and Biological Consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef]
- Pollak, N.; Dölle, C.; Ziegler, M. The power to reduce: Pyridine nucleotides—Small molecules with a multitude of functions. Biochem. J. 2007, 402, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Van Noorden, C.J.F.; Butcher, R.G. A quantitative histochemical study of NADPH-ferrihemoprotein reductase activity. Histochem. J. 1986, 18, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Zangar, R.C. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol. 2004, 199, 316–331. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [Green Version]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [PubMed]
- Holmgren, A.; Lu, J. Thioredoxin and thioredoxin reductase: Current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010, 396, 120–124. [Google Scholar] [CrossRef]
- Kirkman, H.N.; Galiano, S.; Gaetani, G.F. The Function of Catalase-bound NADPH. J. Biol. Chem. 1987, 262, 660–666. [Google Scholar] [PubMed]
- Markolovic, S.; Wilkins, S.E.; Schofield, C.J. Protein Hydroxylation Catalyzed by 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem. 2015, 290, 20712–20722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loenarz, C.; Schofield, C.J. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Chem. Biol. 2008, 4, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Jochmanova, I.; Pacak, K. Pheochromocytoma: The First Metabolic Endocrine Cancer. Clin. Cancer Res. 2016, 22, 5001–5011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagné, L.M.; Boulay, K.; Topisirovic, I.; Huot, M.-É.; Mallette, F.A. Oncogenic Activities of IDH1/2 Mutations: From Epigenetics to Cellular Signaling. Trends Cell Biol. 2017, 27, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1,010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Pansuriya, T.C.; van Eijk, R.; d’Adamo, P.; van Ruler, M.A.J.H.; Kuijjer, M.L.; Oosting, J.; Cleton-Jansen, A.-M.; van Oosterwijk, J.G.; Verbeke, S.L.J.; Meijer, D.; et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat. Genet. 2011, 43, 1256–1261. [Google Scholar] [CrossRef]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Krenacs, L.; Schaerli, P.; Kis, G.; Bagdi, E. Phenotype of neoplastic cells in angioimmunoblastic T-cell lymphoma is consistent with activated follicular B helper T cells. Blood 2006, 108, 1110–1111. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, R.J.; Thota, S.; Nagata, Y.; Patel, B.; Clemente, M.; Przychodzen, B.; Hirsh, C.; Viny, A.D.; Hosano, N.; Bleeker, F.E.; et al. Clinical and biological implications of ancestral and non-ancestral IDH1 and IDH2 mutations in myeloid neoplasms. Leukemia 2015, 29, 2134–2142. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Miyamoto, M.; Sasajima, Y.; Yamazaki, N. Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am. J. Pathol. 2011, 178, 1395–1402. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jabbour, E.; Ravandi, F.; Takahashi, K.; Daver, N.; Routbort, M.; Patel, K.P.; Brandt, M.; Pierce, S.; Kantarjian, H.; et al. IDH1 and IDH2 mutations in myelodysplastic syndromes and role in disease progression. Leukemia 2016, 30, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Mai, M.; McClure, R.F.; Tefferi, A. IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia 2010, 24, 1146–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Kato, Y.; Kaneko, M.K.; Sugawara, M.; Ogasawara, S.; Tsujimoto, Y.; Naganuma, Y.; Yamakawa, M.; Tsuchiya, T.; Takagi, M. Isocitrate dehydrogenase 2 mutation is a frequent event in osteosarcoma detected by a multi-specific monoclonal antibody MsMab-1. Cancer Med. 2013, 2, 803–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiam, A.F.; Cairns, R.A.; Thoms, J.; Dal Pra, A.; Ahmed, O.; Meng, A.; Mak, T.W.; Bristow, R.G. IDH mutation status in prostate cancer. Oncogene 2012, 31, 3826. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Bojdani, E.; Xing, M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem. Biophys. Res. Commun. 2010, 393, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Seo, S.I.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int. J. Cancer 2009, 125, 353–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ye, D.; Guan, K.-L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [Green Version]
- Al-Khallaf, H. Isocitrate dehydrogenases in physiology and cancer: Biochemical and molecular insight. Cell Biosci. 2017, 7, 37. [Google Scholar] [CrossRef]
- Ye, D.; Guan, K.-L.; Xiong, Y. Metabolism, Activity, and Targeting of D- and L-2-Hydroxyglutarates. Trends Cancer 2018, 4, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Enasidenib: First Global Approval. Drugs 2017, 77, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D. Ivosidenib in IDH1 -Mutated Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 379, 1186. [Google Scholar]
- Amary, M.F.; Damato, S.; Halai, D.; Eskandarpour, M.; Berisha, F.; Bonar, F.; McCarthy, S.; Fantin, V.R.; Straley, K.S.; Lobo, S.; et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet. 2011, 43, 1262–1265. [Google Scholar] [CrossRef]
- Hirata, M.; Sasaki, M.; Cairns, R.A.; Inoue, S.; Puviindran, V.; Li, W.Y.; Snow, B.E.; Jones, L.D.; Wei, Q.; Sato, S.; et al. Mutant IDH is sufficient to initiate enchondromatosis in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 2829–2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pansuriya, T.C.; Kroon, H.M.; Bovée, J.V.M.G. Enchondromatosis: Insights on the different subtypes. Int. J. Clin. Exp. Pathol. 2010, 3, 557–569. [Google Scholar]
- Jin, Y.; Elalaf, H.; Watanabe, M.; Tamaki, S.; Hineno, S.; Matsunaga, K.; Woltjen, K.; Kobayashi, Y.; Nagata, S.; Ikeya, M.; et al. Mutant IDH1 Dysregulates the Differentiation of Mesenchymal Stem Cells in Association with Gene-Specific Histone Modifications to Cartilage- and Bone-Related Genes. PLoS ONE 2015, 10, e0131998. [Google Scholar] [CrossRef]
- Kranendijk, M.; Struys, E.A.; van Schaftingen, E.; Gibson, K.M.; Kanhai, W.A.; van der Knaap, M.S.; Amiel, J.; Buist, N.R.; Das, A.M.; de Klerk, J.B.; et al. IDH2 Mutations in Patients with d-2-Hydroxyglutaric Aciduria. Science 2010, 330, 336. [Google Scholar] [CrossRef] [PubMed]
- Van der Knaap, M.S.; Jakobs, C.; Hoffmann, G.F.; Duran, M.; Muntau, A.C.; Schweitzer, S.; Kelley, R.I.; Parrot-Roulaud, F.; Amiel, J.; De Lonlay, P.; et al. D-2-hydroxyglutaric aciduria: Further clinical delineation. J. Inherit. Metab. Dis. 1999, 22, 404–413. [Google Scholar] [CrossRef]
- Kranendijk, M.; Struys, E.A.; Gibson, K.M.; Wickenhagen, W.V.; Abdenur, J.E.; Buechner, J.; Christensen, E.; de Kremer, R.D.; Errami, A.; Gissen, P.; et al. Evidence for genetic heterogeneity in D-2-hydroxyglutaric aciduria. Hum. Mutat. 2010, 31, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Struys, E.A.; Salomons, G.S.; Achouri, Y.; Van Schaftingen, E.; Grosso, S.; Craigen, W.J.; Verhoeven, N.M.; Jakobs, C. Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2005, 76, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Nota, B.; Hamilton, E.M.; Sie, D.; Ozturk, S.; van Dooren, S.J.M.; Ojeda, M.R.F.; Jakobs, C.; Christensen, E.; Kirk, E.P.; Sykut-Cegielska, J.; et al. Novel cases of D-2-hydroxyglutaric aciduria with IDH1 or IDH2 mosaic mutations identified by amplicon deep sequencing. J. Med. Genet. 2013, 50, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Moslehi, J.; Christensen, C.L.; Saha, S.; Tchaicha, J.H.; Ramkissoon, S.H.; Stewart, K.M.; Carretero, J.; Kikuchi, E.; Zhang, H.; et al. D-2-hydroxyglutarate produced by mutant IDH2 causes cardiomyopathy and neurodegeneration in mice. Genes Dev. 2014, 28, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Vissers, L.E.L.M.; Fano, V.; Martinelli, D.; Campos-Xavier, B.; Barbuti, D.; Cho, T.-J.; Dursun, A.; Kim, O.H.; Lee, S.H.; Timpani, G.; et al. Whole-exome sequencing detects somatic mutations of IDH1 in metaphyseal chondromatosis with D-2-hydroxyglutaric aciduria (MC-HGA). Am. J. Med. Genet. Part A 2011, 155, 2609–2616. [Google Scholar] [CrossRef] [Green Version]
- Pierrache, L.H.M.; Kimchi, A.; Ratnapriya, R.; Roberts, L.; Astuti, G.D.N.; Obolensky, A.; Beryozkin, A.; Tjon-Fo-Sang, M.J.H.; Schuil, J.; Klaver, C.C.W.; et al. Whole-Exome Sequencing Identifies Biallelic IDH3A Variants as a Cause of Retinitis Pigmentosa Accompanied by Pseudocoloboma. Ophthalmology 2017, 124, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Dange, M.; McGee, T.L.; Berson, E.L.; Dryja, T.P.; Colman, R.F. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat. Genet. 2008, 40, 1230–1234. [Google Scholar] [CrossRef]
- Robbins, D.; Wittwer, J.A.; Codarin, S.; Circu, M.L.; Aw, T.Y.; Huang, T.-T.; Van Remmen, H.; Richardson, A.; Wang, D.B.; Witt, S.N.; et al. Isocitrate dehydrogenase 1 is downregulated during early skin tumorigenesis which can be inhibited by overexpression of manganese superoxide dismutase. Cancer Sci. 2012, 103, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, F.; Jiang, Y.; Sun, N.; Chen, Z.; Lv, Y.; Shao, K.; Li, N.; Qiu, B.; Gao, Y.; Li, B.; et al. Identification of isocitrate dehydrogenase 1 as a potential diagnostic and prognostic biomarker for non-small cell lung cancer by proteomic analysis. Mol. Cell. Proteom. 2012, 11, M111.008821. [Google Scholar] [CrossRef]
- Al-Amodi, H.S.A.B.; Nabih, E.S.; Kamel, H.F.M.; El Sayed, M.A.; Dwedar, I.A.M. Wild-Type Isocitrate Dehydrogenase 1 Over-Expression is Related to Cancer Stem Cells Survival in Lung Adenocarcinoma. Cancer Investig. 2018, 36, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; He, Y.; Tan, Z.; Lu, J.; Li, L.; Song, X.; Shi, F.; Xie, L.; You, S.; Luo, X.; et al. Wild-type IDH2 promotes the Warburg effect and tumor growth through HIF1α in lung cancer. Theranostics 2018, 8, 4050–4061. [Google Scholar] [CrossRef]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Chen, Z.; Tan, F.; Zhang, B.; Yao, R.; Zhou, C.; Li, J.; Gao, Y.; Liu, Z.; Tan, X.; et al. Isocitrate dehydrogenase 1 is a novel plasma biomarker for the diagnosis of non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 5136–5145. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, Y.; Chen, J.; Qiu, J.; Huang, K.; Wu, M.; Xia, C. IDH1 R132H Mutation Enhances Cell Migration by Activating AKT-mTOR Signaling Pathway, but Sensitizes Cells to 5-FU Treatment as NADPH and GSH Are Reduced. PLoS ONE 2017, 12, e0169038. [Google Scholar] [CrossRef]
- Ma, Q.-L.; Wang, J.-H.; Wang, Y.-G.; Hu, C.; Mu, Q.-T.; Yu, M.-X.; Wang, L.; Wang, D.-M.; Yang, M.; Yin, X.-F.; et al. High IDH1 expression is associated with a poor prognosis in cytogenetically normal acute myeloid leukemia. Int. J. Cancer 2015, 137, 1058–1065. [Google Scholar] [CrossRef]
- Zarei, M.; Lal, S.; Parker, S.J.; Nevler, A.; Vaziri-Gohar, A.; Dukleska, K.; Mambelli-Lisboa, N.C.; Moffat, C.; Blanco, F.F.; Chand, S.N.; et al. Posttranscriptional Upregulation of IDH1 by HuR Establishes a Powerful Survival Phenotype in Pancreatic Cancer Cells. Cancer Res. 2017, 77, 4460–4471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef]
- Liu, W.-R.; Tian, M.-X.; Jin, L.; Yang, L.-X.; Ding, Z.-B.; Shen, Y.-H.; Peng, Y.-F.; Zhou, J.; Qiu, S.-J.; Dai, Z.; et al. High expression of 5-hydroxymethylcytosine and isocitrate dehydrogenase 2 is associated with favorable prognosis after curative resection of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2014, 33, 32. [Google Scholar] [CrossRef]
- Tian, G.-Y.; Zang, S.-F.; Wang, L.; Luo, Y.; Shi, J.-P.; Lou, G.-Q. Isocitrate Dehydrogenase 2 Suppresses the Invasion of Hepatocellular Carcinoma Cells via Matrix Metalloproteinase 9. Cell. Physiol. Biochem. 2015, 37, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Wu, D. Isocitrate dehydrogenase 2 inhibits gastric cancer cell invasion via matrix metalloproteinase 7. Tumor Biol. 2016, 37, 5225–5230. [Google Scholar] [CrossRef]
- Tanaka, H.; Sasayama, T.; Tanaka, K.; Nakamizo, S.; Nishihara, M.; Mizukawa, K.; Kohta, M.; Koyama, J.; Miyake, S.; Taniguchi, M.; et al. MicroRNA-183 upregulates HIF-1α by targeting isocitrate dehydrogenase 2 (IDH2) in glioma cells. J. Neurooncol. 2013, 111, 273–283. [Google Scholar] [CrossRef]
- Wang, X.-J.; Zhang, D.-L.; Fu, C.; Wei, B.-Z.; Li, G.-J. MiR-183 modulates multi-drug resistance in hepatocellular cancer (HCC) cells via miR-183-IDH2/SOCS6-HIF-1α feedback loop. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2020–2027. [Google Scholar] [PubMed]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef]
- Yu, W.; Denu, R.A.; Krautkramer, K.A.; Grindle, K.M.; Yang, D.T.; Asimakopoulos, F.; Hematti, P.; Denu, J.M. Loss of SIRT3 Provides Growth Advantage for B Cell Malignancies. J. Biol. Chem. 2016, 291, 3268–3279. [Google Scholar] [CrossRef]
- Chen, X.; Xu, W.; Wang, C.; Liu, F.; Guan, S.; Sun, Y.; Wang, X.; An, D.; Wen, Z.; Chen, P.; et al. The clinical significance of isocitrate dehydrogenase 2 in esophageal squamous cell carcinoma. Am. J. Cancer Res. 2017, 7, 700–714. [Google Scholar] [PubMed]
- Park, J.H.; Ku, H.J.; Lee, J.H.; Park, J.-W. Idh2 Deficiency Exacerbates Acrolein-Induced Lung Injury through Mitochondrial Redox Environment Deterioration. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Wang, L.-N.; Tong, S.-W.; Hu, H.-D.; Ye, F.; Li, S.-L.; Ren, H.; Zhang, D.-Z.; Xiang, R.; Yang, Y.-X. Quantitative proteome analysis of ovarian cancer tissues using a iTRAQ approach. J. Cell. Biochem. 2012, 113, 3762–3772. [Google Scholar] [CrossRef] [PubMed]
- Guirguis, A.; Elishaev, E.; Oh, S.-H.; Tseng, G.C.; Zorn, K.; DeLoia, J.A. Use of gene expression profiles to stage concurrent endometrioid tumors of the endometrium and ovary. Gynecol. Oncol. 2008, 108, 370–376. [Google Scholar] [CrossRef]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhu, Y.; Park, S.-H.; Liu, G.; O’Brien, J.; Jiang, H.; Gius, D. SIRT3-Mediated Dimerization of IDH2 Directs Cancer Cell Metabolism and Tumor Growth. Cancer Res. 2017, 77, 3990–3999. [Google Scholar] [CrossRef]
- Torrens-Mas, M.; Pons, D.G.; Sastre-Serra, J.; Oliver, J.; Roca, P. SIRT3 Silencing Sensitizes Breast Cancer Cells to Cytotoxic Treatments Through an Increment in ROS Production. J. Cell. Biochem. 2017, 118, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Xing, S.; Li, Z.; Li, J.; Gong, P.; Xu, X.; Chang, L.; Jin, X.; Gao, F.; Li, W.; et al. Altered expression levels of IDH2 are involved in the development of colon cancer. Exp. Ther. Med. 2012, 4, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koseki, J.; Colvin, H.; Fukusumi, T.; Nishida, N.; Konno, M.; Kawamoto, K.; Tsunekuni, K.; Matsui, H.; Doki, Y.; Mori, M.; et al. Mathematical analysis predicts imbalanced IDH1/2 expression associates with 2-HG-inactivating β-oxygenation pathway in colorectal cancer. Int. J. Oncol. 2015, 46, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, S.Y.; Ku, H.J.; Jeon, Y.H.; Lee, H.W.; Lee, J.; Kwon, T.K.; Park, K.M.; Park, J.-W. Suppression of tumorigenesis in mitochondrial NADP+-dependent isocitrate dehydrogenase knock-out mice. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Morinibu, A.; Kobayashi, M.; Zhu, Y.; Wang, X.; Goto, Y.; Yeom, C.J.; Zhao, T.; Hirota, K.; Shinomiya, K.; et al. Aberrant IDH3α expression promotes malignant tumor growth by inducing HIF-1-mediated metabolic reprogramming and angiogenesis. Oncogene 2015, 34, 4758–4766. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Kotake, Y.; Demizu, Y.; Kurihara, M.; Sekino, Y.; Kanda, Y. NAD-dependent isocitrate dehydrogenase as a novel target of tributyltin in human embryonic carcinoma cells. Sci. Rep. 2014, 4, 5952. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell. 2009, 136, 823–837. [Google Scholar] [CrossRef]
- Mohrenz, I.V.; Antonietti, P.; Pusch, S.; Capper, D.; Balss, J.; Voigt, S.; Weissert, S.; Mukrowsky, A.; Frank, J.; Senft, C.; et al. Isocitrate dehydrogenase 1 mutant R132H sensitizes glioma cells to BCNU-induced oxidative stress and cell death. Apoptosis 2013, 18, 1416–1425. [Google Scholar] [CrossRef]
- Li, S.; Chou, A.P.; Chen, W.; Chen, R.; Deng, Y.; Phillips, H.S.; Selfridge, J.; Zurayk, M.; Lou, J.J.; Everson, R.G.; et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro-Oncology 2013, 15, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-B.; Dong, D.-F.; Wang, M.-D.; Gao, K. IDH1 overexpression induced chemotherapy resistance and IDH1 mutation enhanced chemotherapy sensitivity in Glioma cells in vitro and in vivo. Asian Pac. J. Cancer Prev. 2014, 15, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Spitz, D.R.; Azzam, E.I.; Jian Li, J.; Gius, D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: A unifying concept in stress response biology. Cancer Metastasis Rev. 2004, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.-H.; Lee, S.-H.; Suk Chun, H.; Min Lee, S.; Koh, H.-J.; Lee, S.-E.; Chun, J.-S.; Park, J.-W.; Huh, T.-L. Cellular Defense against UVB-Induced Phototoxicity by Cytosolic NADP+-Dependent Isocitrate Dehydrogenase. Biochem. Biophys. Res. Commun. 2002, 292, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Wahl, D.R.; Dresser, J.; Wilder-Romans, K.; Parsels, J.D.; Zhao, S.G.; Davis, M.; Zhao, L.; Kachman, M.; Wernisch, S.; Burant, C.F.; et al. Glioblastoma Therapy Can Be Augmented by Targeting IDH1-Mediated NADPH Biosynthesis. Cancer Res. 2017, 77, 960–970. [Google Scholar] [CrossRef]
- Kim, S.Y.; Yoo, Y.H.; Park, J.-W. Silencing of mitochondrial NADP(+)-dependent isocitrate dehydrogenase gene enhances glioma radiosensitivity. Biochem. Biophys. Res. Commun. 2013, 433, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Van Straten, D.; Mashayekhi, V.; de Bruijn, H.S.; Oliveira, S.; Robinson, D.J. Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 2017, 9, 19. [Google Scholar] [CrossRef]
- Kim, S.Y.; Lee, S.M.; Tak, J.K.; Choi, K.S.; Kwon, T.K.; Park, J.-W. Regulation of singlet oxygen-induced apoptosis by cytosolic NADP+-dependent isocitrate dehydrogenase. Mol. Cell. Biochem. 2007, 302, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2017, 17, 183–196. [Google Scholar] [CrossRef]
- Bergaggio, E.; Riganti, C.; Garaffo, G.; Vitale, N.; Mereu, E.; Bandini, C.; Pellegrino, E.; Pullano, V.; Omedè, P.; Todoerti, K.; et al. IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood 2019, 133, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Cagnetta, A.; Cea, M.; Calimeri, T.; Acharya, C.; Fulciniti, M.; Tai, Y.-T.; Hideshima, T.; Chauhan, D.; Zhong, M.Y.; Patrone, F.; et al. Intracellular NAD+ depletion enhances bortezomib-induced anti-myeloma activity. Blood 2013, 122, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Yao, Y.; Xu, H.-Z.; Qin, Z.-Y. Isocitrate Dehydrogenase (IDH)1/2 Mutations as Prognostic Markers in Patients with Glioblastomas. Medicine (Baltimore) 2016, 95, e2583. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Krönke, J.; Bullinger, L.; Späth, D.; Kayser, S.; Zucknick, M.; Götze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef]
- Xue, Y.; Chen, Q.; Ding, T.; Sun, J. SiO2 nanoparticle-induced impairment of mitochondrial energy metabolism in hepatocytes directly and through a Kupffer cell-mediated pathway in vitro. Int. J. Nanomed. 2014, 9, 2891. [Google Scholar]
- Planchon, M.; Léger, T.; Spalla, O.; Huber, G.; Ferrari, R. Metabolomic and proteomic investigations of impacts of titanium dioxide nanoparticles on Escherichia coli. PLoS ONE 2017, 12, e0178437. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| IDH1 mut. | IDH2 mut. | IDH3 mut. |
|---|---|---|
| Ollier disease [24,47,50] | Ollier disease [24,47] | Retinitis pigmentosa [59,60] |
| Maffucci syndrome [24,47,50] | Maffucci syndrome [47] | |
| Spondyloenchondromatosis with D-2-hydroxyglutaric aciduria [55,57] | D-2-hydroxyglutaric aciduria [51,55] |
| IDH1 Levels | ||
| Downregulated | Overexpressed | Not Specified |
| Early skin tumorigenesis (OS) [61] | Lung squamous cell carcinoma (OG) [62,65,66] | Acute myeloid leukemia (OG) [68] |
| Lung adenocarcinoma (OG) [62,63,65,66] | ||
| Primary glioblastoma (OG) [65,67] | ||
| Angioimmunoblastic lymphoma [65] | ||
| Anaplastic large cell lymphoma [65] | ||
| Peripheral T cell lymphoma [65] | ||
| Diffuse large B cell lymphoma (OG) [65] | ||
| Pancreatic ductal adenocarcinoma (OG) [69] | ||
| Imbalance of IDH1/2 in colorectal cancer (OG) [86] | ||
| IDH2 Levels | ||
| Downregulated | Overexpressed | Not Specified |
| Melanoma (OS) [70] | Esophageal squamous cell cancer (OG) [78] | Lewis lung carcinoma (OS) [79] |
| Kidney cancer [64] | Lung cancer (OG) [64] | Pancreatic cancer (OG) [64] |
| Hepatocellular carcinoma (OS) [71,72] | Ovarian cancer [64,80] | |
| Gastric cancer (OS) [64,73] | Endometroid carcinomas of the endometrium vs. endometroid carcinomas of the ovary [81] | |
| Glioblastoma [75] | Prostate cancer [82] | |
| Grade III glioma [75] | Testis cancer [82] | |
| Mantle cell lymphoma (OS) [77] | Eye cancer [82] | |
| Chronic lymphocytic leukemia [77] | Nervous cancer [82] | |
| Acute lymphocytic leukemia [77] | Breast cancer (OG/OS) [64,82,83,84] | |
| Burkitt’s lymphoma (OS) [77] | Infiltrating colorectal cancer (OG) [85] | |
| High-risk luminal B vs. low-risk luminal A breast cancer [83] | ||
| In situ colorectal cancer [85] | ||
| Imbalance of IDH1/2 in colorectal cancer (OG) [86] | ||
| IDH3 Levels | ||
| Downregulated | Overexpressed | Not Specified |
| Lung cancer (OG) [88] | ||
| Breast cancer (OG) [88] | ||
| Cervical adenocarcinoma (OG) [88] | ||
| Embryonic carcinoma (OG) [89] | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergaggio, E.; Piva, R. Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy. Cancers 2019, 11, 563. https://doi.org/10.3390/cancers11040563
Bergaggio E, Piva R. Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy. Cancers. 2019; 11(4):563. https://doi.org/10.3390/cancers11040563
Chicago/Turabian StyleBergaggio, Elisa, and Roberto Piva. 2019. "Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy" Cancers 11, no. 4: 563. https://doi.org/10.3390/cancers11040563
APA StyleBergaggio, E., & Piva, R. (2019). Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy. Cancers, 11(4), 563. https://doi.org/10.3390/cancers11040563