Dasatinib Treatment Increases Sensitivity to c-Met Inhibition in Triple-Negative Breast Cancer Cells

 ,
,  , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Proliferation Assays
2.3. Doubling Time Assays
2.4. Short-Term Resistance Assay
2.5. Invasion/Migration Assays
2.6. Dasatinib Accumulation Assays
2.7. Protein Extraction and Western Blotting
2.8. Luminex Magnetic Bead Assays
2.9. DNA Extraction and Nested PCR Amplification of Src Exons 9–12
2.10. Cycle Sequencing of PCR Products
2.11. Statistical Analysis
3. Results
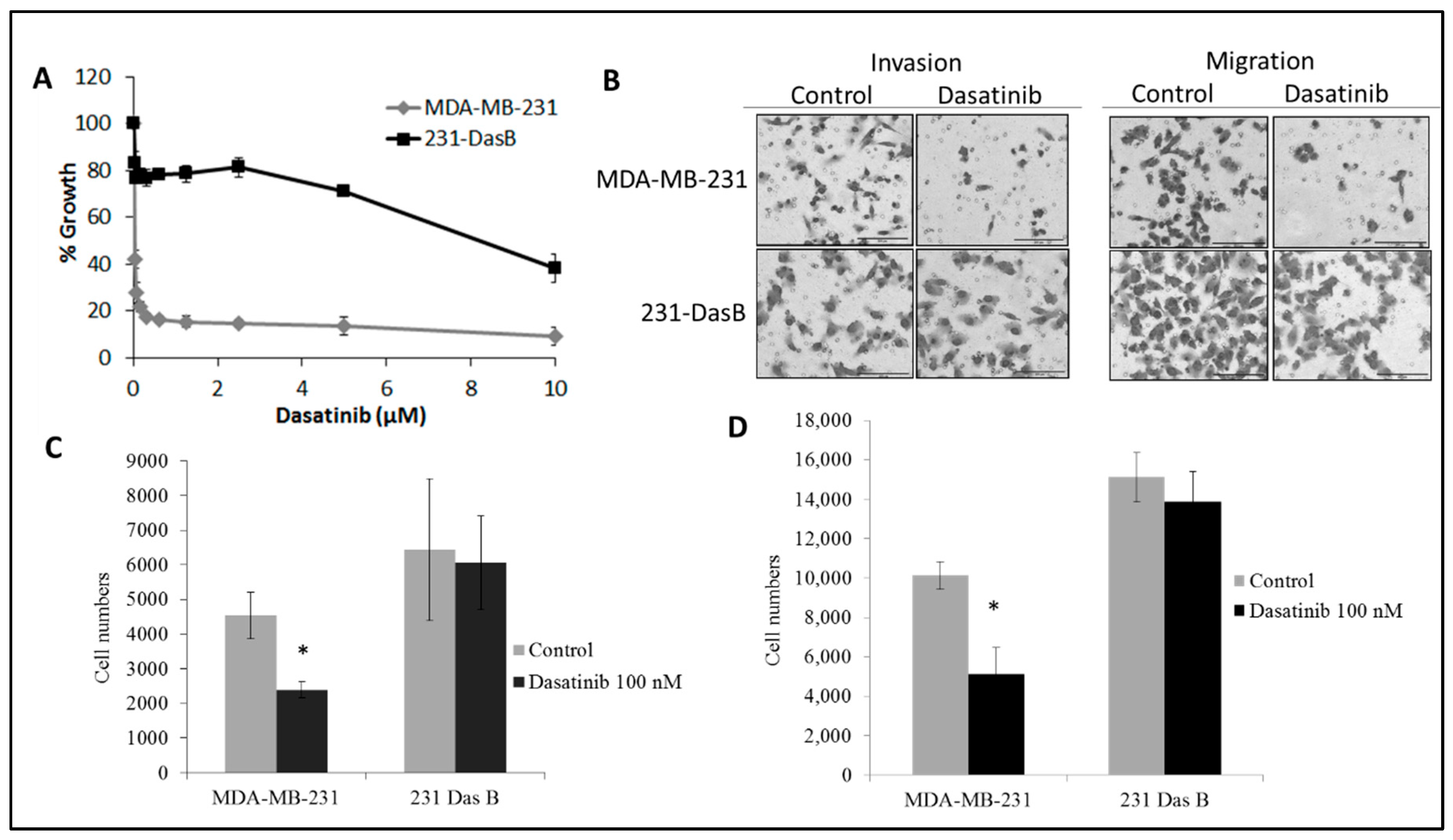
3.1. Dasatinib Exposure Induces a Resistant Phenotype
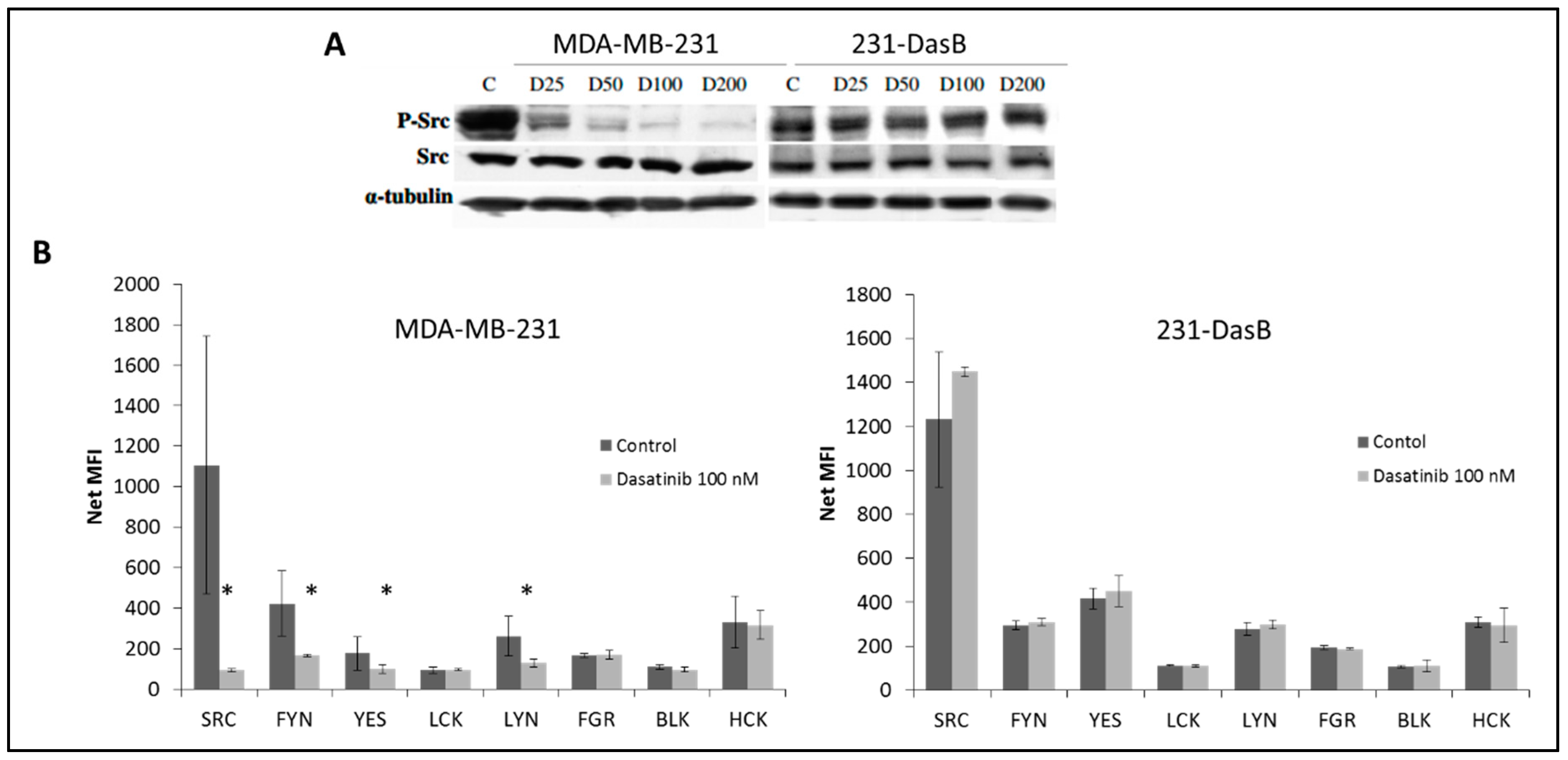
3.2. Phosphorylation of Src Is Altered in Dasatinib-Resistant Cells
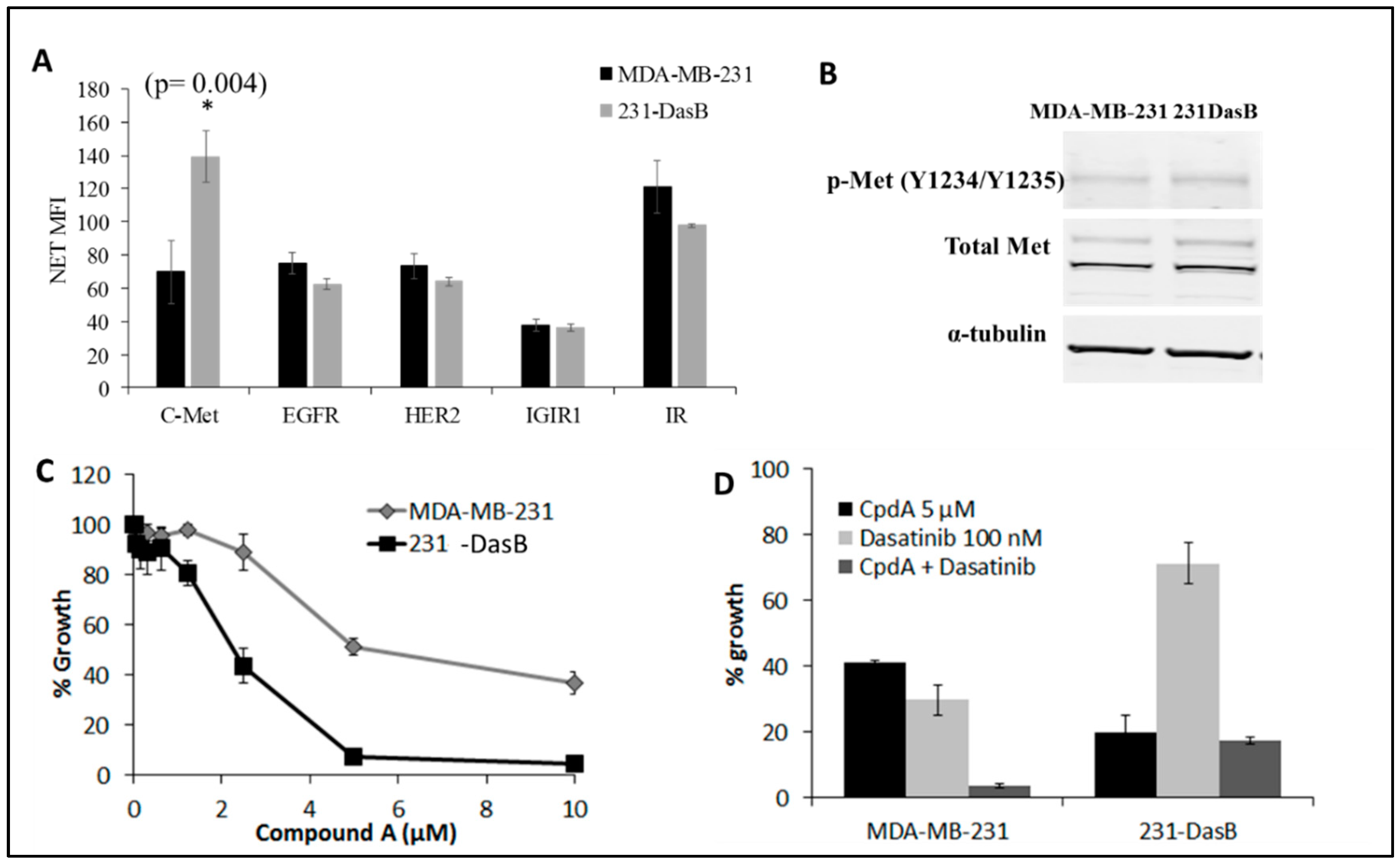
3.3. cMet Signalling Is Increased in Dasatinib-Resistant Cells
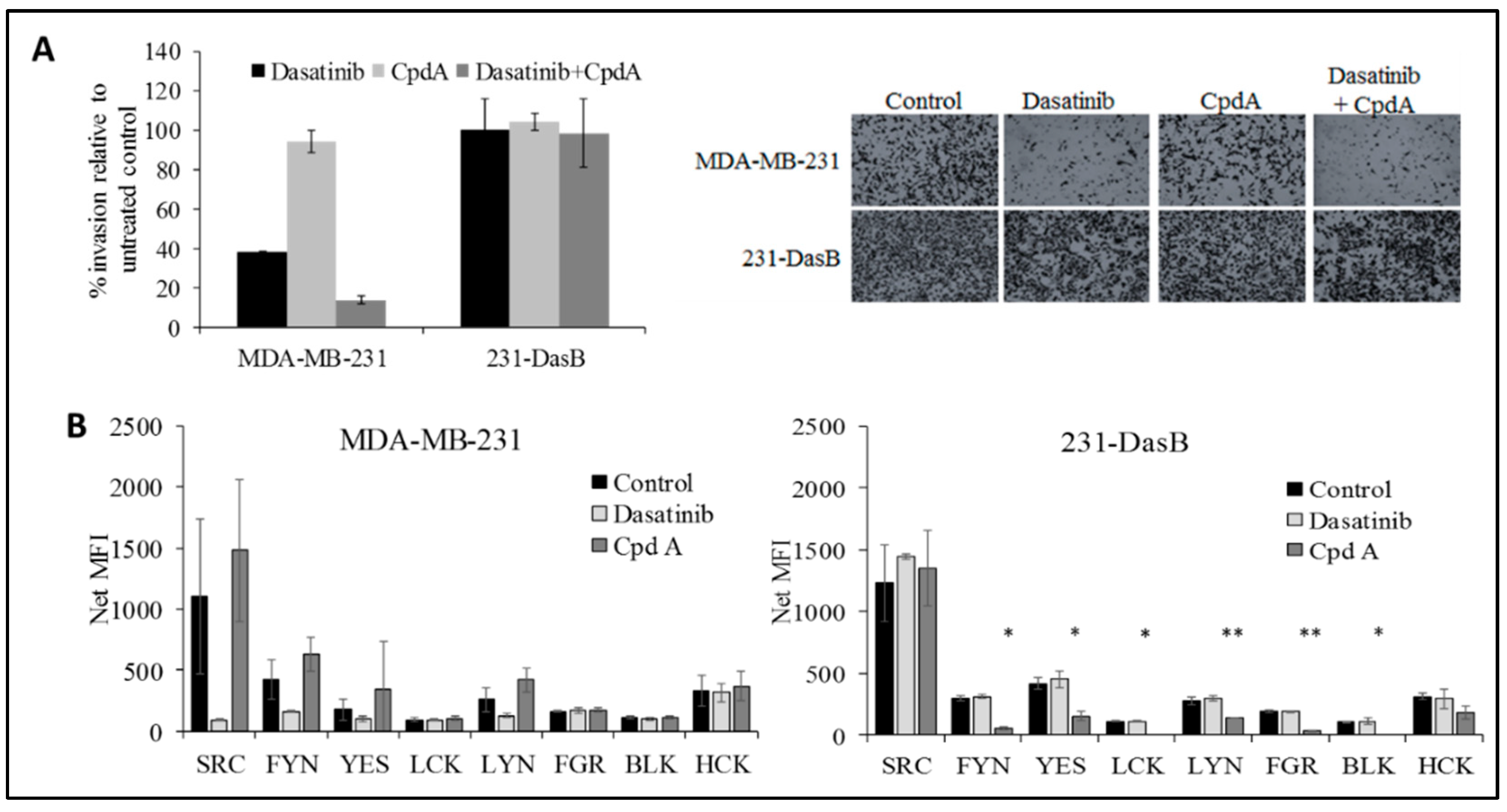
3.4. cMET Inhibition Blocks Dasatinib Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pogoda, K.; Niwińska, A.; Murawska, M.; Pieńkowski, T. Analysis of pattern, time and risk factors influencing recurrence in triple-negative breast cancer patients. Med. Oncol. 2013, 30, 388. [Google Scholar] [CrossRef]
- Narod, S.A.; Dent, R.A.; Foulkes, W.D. CCR 20thanniversary commentary: Triple-negative breast cancer in 2015—Still in the ballpark. Clin. Cancer Res. 2015, 21, 3813–3814. [Google Scholar] [CrossRef]
- Tryfonopoulos, D.; Walsh, S.; Collins, D.M.; Flanagan, L.; Quinn, C.; Corkery, B.; McDermott, E.W.; Evoy, D.; Pierce, A.; O’Donovan, N.; et al. Src: A potential target for the treatment of triple-negative breast cancer. Ann. Oncol. 2011, 22, 2234–2240. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef]
- Kim, M.P.; Park, S.I.; Kopetz, S.; Gallick, G.E. Src family kinases as mediators of endothelial permeability: Effects on inflammation and metastasis. Cell Tissue Res. 2009, 335, 249. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Iida, M.; Kruser, T.J.; Nechrebecki, M.M.; Dunn, E.F.; Armstrong, E.A.; Huang, S.; Harari, P.M. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol. Ther. 2009, 8, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S. Targeting Src in breast cancer. Ann. Oncol. 2008, 19, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Dering, J.; Ginther, C.; Wilson, C.A.; Glaspy, P.; Tchekmedyian, N.; Slamon, D.J. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/”triple- negative” breast cancer cell lines growing in vitro. Breast Cancer Res. Treat. 2007, 105, 319–326. [Google Scholar] [CrossRef]
- Huang, F.; Reeves, K.; Han, X.; Fairchild, C.; Platero, S.; Wong, T.W.; Lee, F.; Shaw, P.; Clark, E. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: Rationale for patient selection. Cancer Res. 2007, 67, 2226–2238. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Herold, C.I.; Chadaram, V.; Peterson, B.L.; Marcom, P.K.; Hopkins, J.; Kimmick, G.G.; Favaro, J.; Hamilton, E.; Welch, R.A.; Bacus, S.; et al. Phase II trial of dasatinib in patients with metastatic breast cancer using real-time pharmacodynamic tissue biomarkers of Src inhibition to escalate dosing. Clin. Res. 2011, 17, 6061–6070. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.; Bengala, C.; Ibrahim, N.; Strauss, L.; Fairchild, J.; Sy, O.; Roche, H.; Sparano, J.; Goldstein, L. Phase II trial of dasatinib in triple-negative breast cancer: Results of study CA180059. Cancer Res. 2009, 69, 3118. [Google Scholar] [CrossRef]
- Pusztai, L.; Moulder, S.; Altan, M.; Kwiatkowski, D.; Valero, V.; Ueno, N.T.; Esteva, F.J.; Avritscher, R.; Qi, Y.; Strauss, L.; et al. Gene signature-guided dasatinib therapy in metastatic breast cancer. Clin. Cancer Res. 2014, 20, 5265–5271. [Google Scholar] [CrossRef]
- Roche, S.; McMahon, G.; Clynes, M.; O’Connor, R. Development of a high-performance liquid chromatographic-mass spectrometric method for the determination of cellular levels of the tyrosine kinase inhibitors lapatinib and dasatinib. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3982–3990. [Google Scholar] [CrossRef]
- Hiwase, D.K.; Saunders, V.; Hewett, D.; Frede, A.; Zrim, S.; Dang, P.; Eadie, L.; To, L.B.; Melo, J.; Kumar, S.; et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: Therapeutic implications. Clin. Cancer Res. 2008, 14, 3881–3888. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.M.; Crown, J.; O’Donovan, N.; Devery, A.; O’Sullivan, F.; O’Driscoll, L.; Clynes, M.; O’Connor, R. Tyrosine kinase inhibitors potentiate the cytotoxicity of MDR-substrate anticancer agents independent of growth factor receptor status in lung cancer cell lines. Investig. New Drugs 2010, 28, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Maliepaard, M.; Van Gastelen, M.A.; Tohgo, A.; Hausheer, F.H.; Van Waardenburg, R.C.A.M.; De Jong, L.A.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.M. Circumvention of breast cancer resistance protein (BCRP)-mediated resistance to camptothecins in vitro using non-substrate drugs or the BCRP inhibitor GF120918. Clin. Cancer Res. 2001, 7, 935–941. [Google Scholar] [PubMed]
- Hyafil, F.; Vergely, C.; Vignaud, P.; Du Grand-Perret, T. In Vitro and in Vivo Reversal of Multidrug Resistance by GF120918, an Acridonecarboxamide Derivative. Cancer Res. 1993, 53, 4595–4602. [Google Scholar] [PubMed]
- Anderson, S.K.; Gibbs, C.P.; Tanaka, A.; Kung, H.J.; Fujita, D.J. Human Cellular src Gene: Nucleotide Sequence and Derived Amino Acid Sequence of the Region Coding for the Carboxy-Terminal Two-Thirds of pp6Oc-src. Mol. Cell Biol. 1985, 5, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Mao, W.; Coppola, D.; Kang, J.; Loubeau, J.M.; Trudeau, W.; Karl, R.; Fujita, D.J.; Jove, R.; Yeatman, T.J. Activating SRC mutation in a subset of advanced human colon cancers. Nat. Genet. 1999, 21, 187–190. [Google Scholar] [CrossRef]
- Takahashi, S.; Miyazaki, M.; Okamoto, I.; Ito, Y.; Ueda, K.; Seriu, T.; Nakagawa, K.; Hatake, K. Phase I study of dasatinib (BMS-354825) in Japanese patients with solid tumors. Cancer Sci. 2011, 102, 2058–2064. [Google Scholar] [CrossRef]
- Haura, E.B.; Tanvetyanon, T.; Chiappori, A.; Williams, C.; Simon, G.; Antonia, S.; Gray, J.; Litschauer, S.; Tetteh, L.; Neuger, A.; et al. Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 1387–1394. [Google Scholar] [CrossRef]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.S.J.; Browne, B.C.; Conlon, N.T.; O’Brien, N.A.; Slamon, D.J.; Henry, M.; Meleady, P.; Clynes, M.; Dowling, P.; Crown, J.; et al. PP2A inhibition overcomes acquired resistance to HER2 targeted therapy. Mol. Cancer 2014, 13, 157. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, M.; Kobayashi, K.; Sagae, S.; Nishioka, Y.; Ishioka, S.I.; Terasawa, K.; Tokino, T.; Kudo, R. Mutation of the SRC gene in endometrial carcinoma. Jpn. J. Cancer Res. 2000, 91, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Okamoto, I.; Okamoto, W.; Hatashita, E.; Yamada, Y.; Kuwata, K.; Nishio, K.; Fukuoka, M.; Jänne, P.A.; Nakagawa, K. Effects of Src inhibitors on cell growth and epidermal growth factor receptor and MET signaling in gefitinib-resistant non-small cell lung cancer cells with acquired MET amplification. Cancer Sci. 2010, 101, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Nappi, L.; Rosa, R.; Marciano, R.; D’Amato, C.; D’Amato, V.; Damiano, V.; Raimondo, L.; Iommelli, F.; Scorziello, A.; et al. Epidermal growth factor-receptor activation modulates Src-dependent resistance to lapatinib in breast cancer models. Breast Cancer Res. 2014, 16, R45. [Google Scholar] [CrossRef] [PubMed]
- Pinedo-Carpio, E.; Davidson, D.; Marignac, V.L.M.; Panasci, J.; Aloyz, R. Adaptive metabolic rewiring to chronic SFK inhibition. Oncotarget 2017, 8, 66758. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. TGFβ1 rapidly activates Src through a non-canonical redox signaling mechanism. Arch. Biochem. Biophys. 2015, 568, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hage, C.; Rausch, V.; Giese, N.; Giese, T.; Schönsiegel, F.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Herr, I. The novel c-Met inhibitor cabozantinib overcomes gemcitabine resistance and stem cell signaling in pancreatic cancer. Cell Death Dis. 2013, 4, e627. [Google Scholar] [CrossRef]
- Moschetta, M.; Basile, A.; Ferrucci, A.; Frassanito, M.A.; Rao, L.; Ria, R.; Solimando, A.G.; Giuliani, N.; Boccarelli, A.; Fumarola, F.; et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin. Cancer Res. 2013, 19, 4371–4382. [Google Scholar] [CrossRef] [PubMed]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Shawn Carbonell, W.; Hu, Y.L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
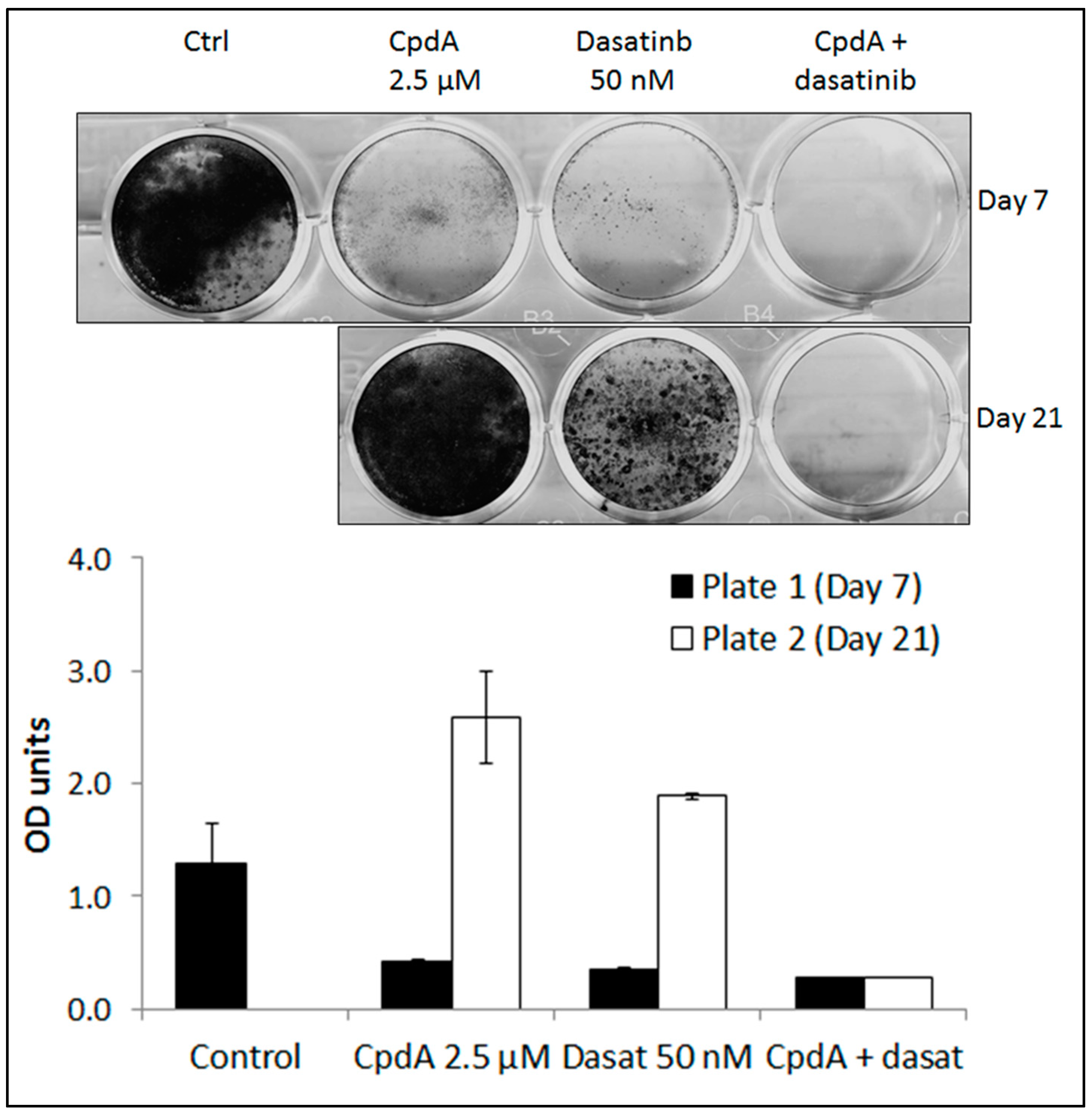{kind=link}
| Cell Line | Control | D 50 nM | D 100 nM |
|---|---|---|---|
| MDA-MB-231 | 17.6 ± 1.2 | 32.2 ± 3.3* | 46.8 ± 5.1* |
| 231 DasB | 19.1 ± 2.4 | 21.0 ± 0.2 | 21.2 ± 3.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaule, P.; Mukherjee, N.; Corkery, B.; Eustace, A.J.; Gately, K.; Roche, S.; O’Connor, R.; O’Byrne, K.J.; Walsh, N.; Duffy, M.J.; et al. Dasatinib Treatment Increases Sensitivity to c-Met Inhibition in Triple-Negative Breast Cancer Cells. Cancers 2019, 11, 548. https://doi.org/10.3390/cancers11040548
Gaule P, Mukherjee N, Corkery B, Eustace AJ, Gately K, Roche S, O’Connor R, O’Byrne KJ, Walsh N, Duffy MJ, et al. Dasatinib Treatment Increases Sensitivity to c-Met Inhibition in Triple-Negative Breast Cancer Cells. Cancers. 2019; 11(4):548. https://doi.org/10.3390/cancers11040548
Chicago/Turabian StyleGaule, Patricia, Nupur Mukherjee, Brendan Corkery, Alex J. Eustace, Kathy Gately, Sandra Roche, Robert O’Connor, Kenneth J. O’Byrne, Naomi Walsh, Michael J. Duffy, and et al. 2019. "Dasatinib Treatment Increases Sensitivity to c-Met Inhibition in Triple-Negative Breast Cancer Cells" Cancers 11, no. 4: 548. https://doi.org/10.3390/cancers11040548
APA StyleGaule, P., Mukherjee, N., Corkery, B., Eustace, A. J., Gately, K., Roche, S., O’Connor, R., O’Byrne, K. J., Walsh, N., Duffy, M. J., Crown, J., & O’Donovan, N. (2019). Dasatinib Treatment Increases Sensitivity to c-Met Inhibition in Triple-Negative Breast Cancer Cells. Cancers, 11(4), 548. https://doi.org/10.3390/cancers11040548