Strategies to Improve Cancer Immune Checkpoint Inhibitors Efficacy, Other Than Abscopal Effect: A Systematic Review


 ,
,  ,
, 
Abstract
1. Introduction
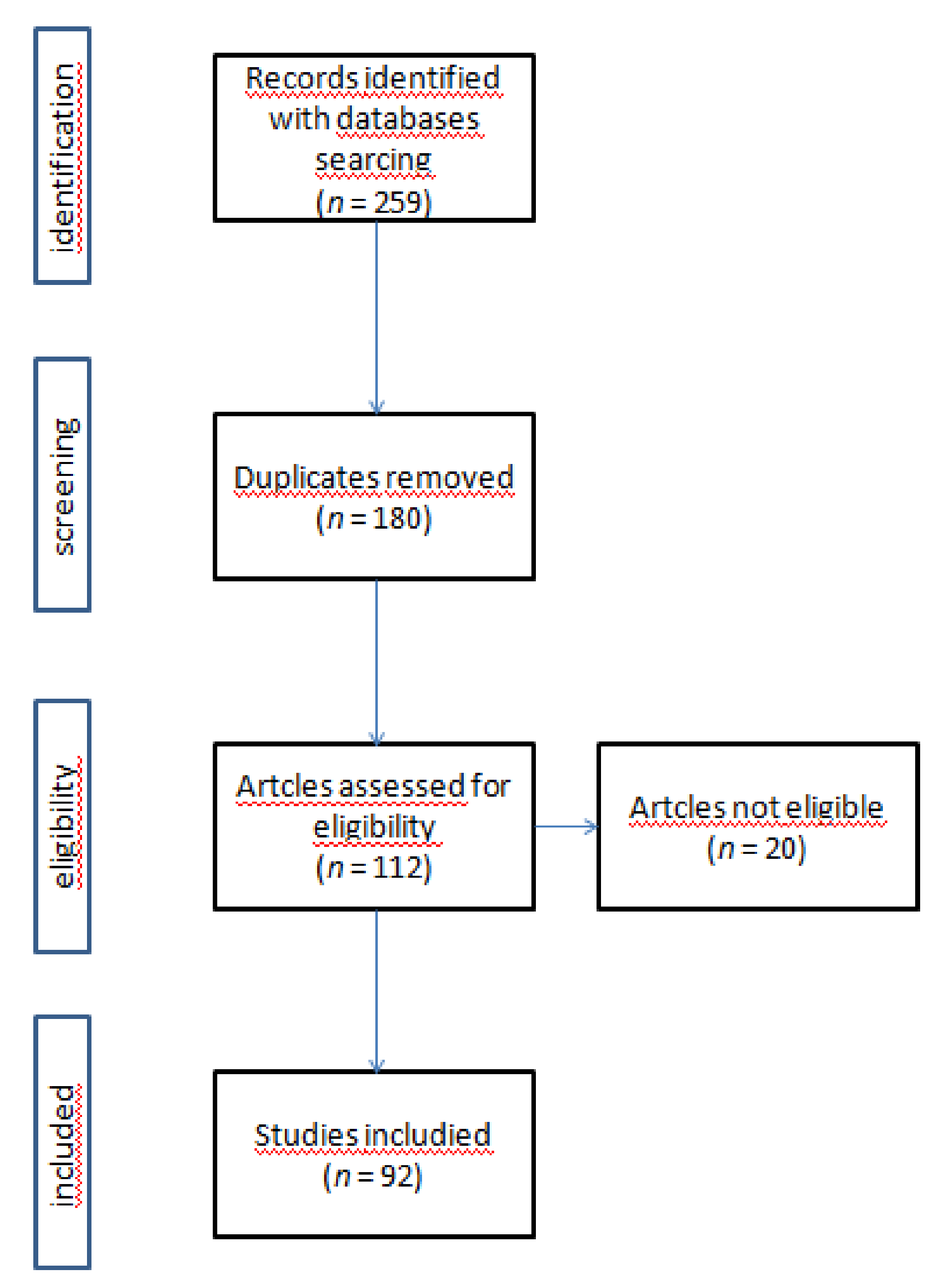
2. Materials and Methods
2.1. Research Strategy
2.2. Inclusion/Exclusion Criteria
2.3. Study Selection
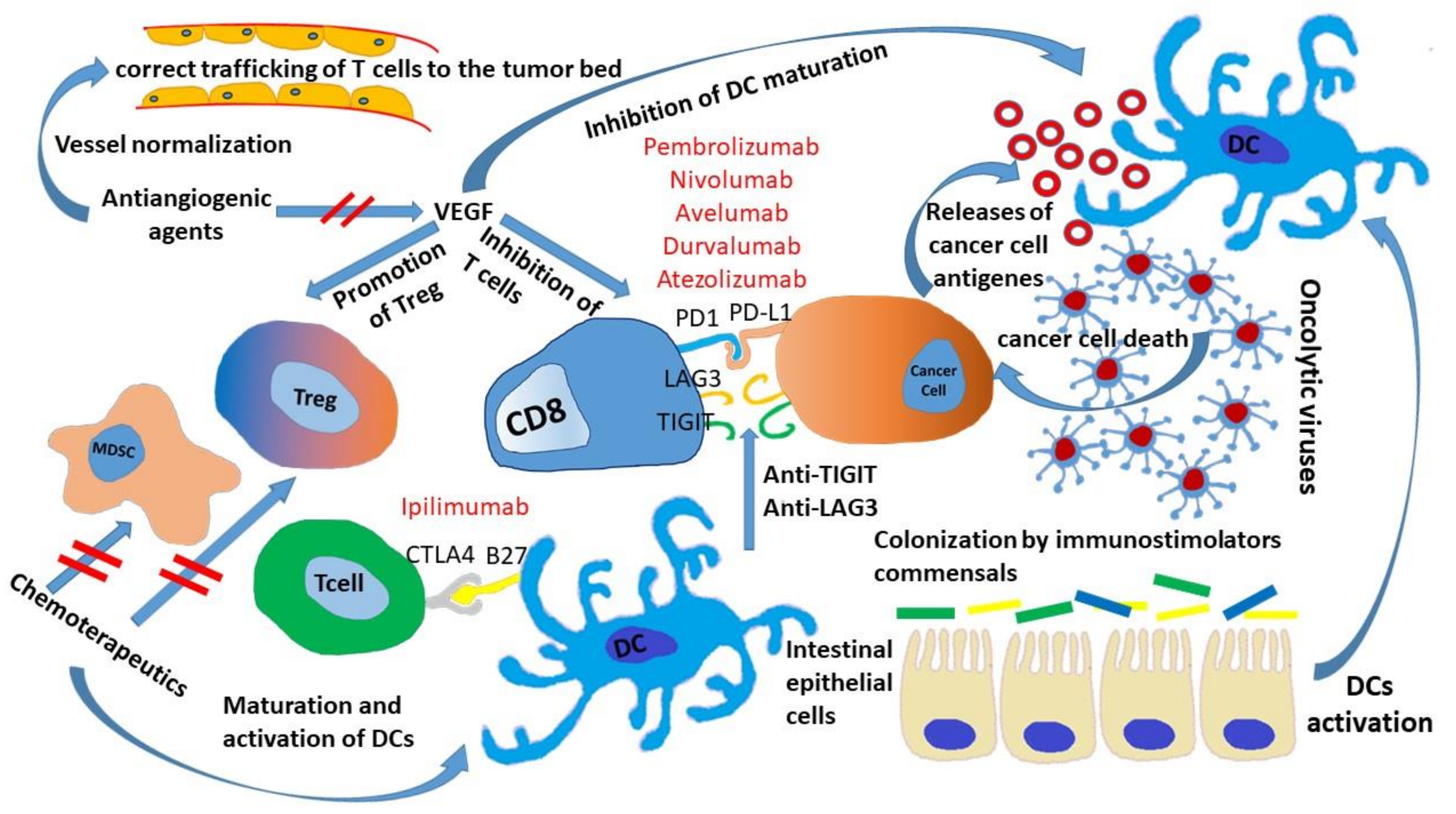
3. Microbiota and ICIs
4. Chemotherapeutics Sensitizing Tumor to ICIs
5. ICIs and Antiangiogenic Drugs
6. Strategies Involving Other Co-Inhibitor Receptors
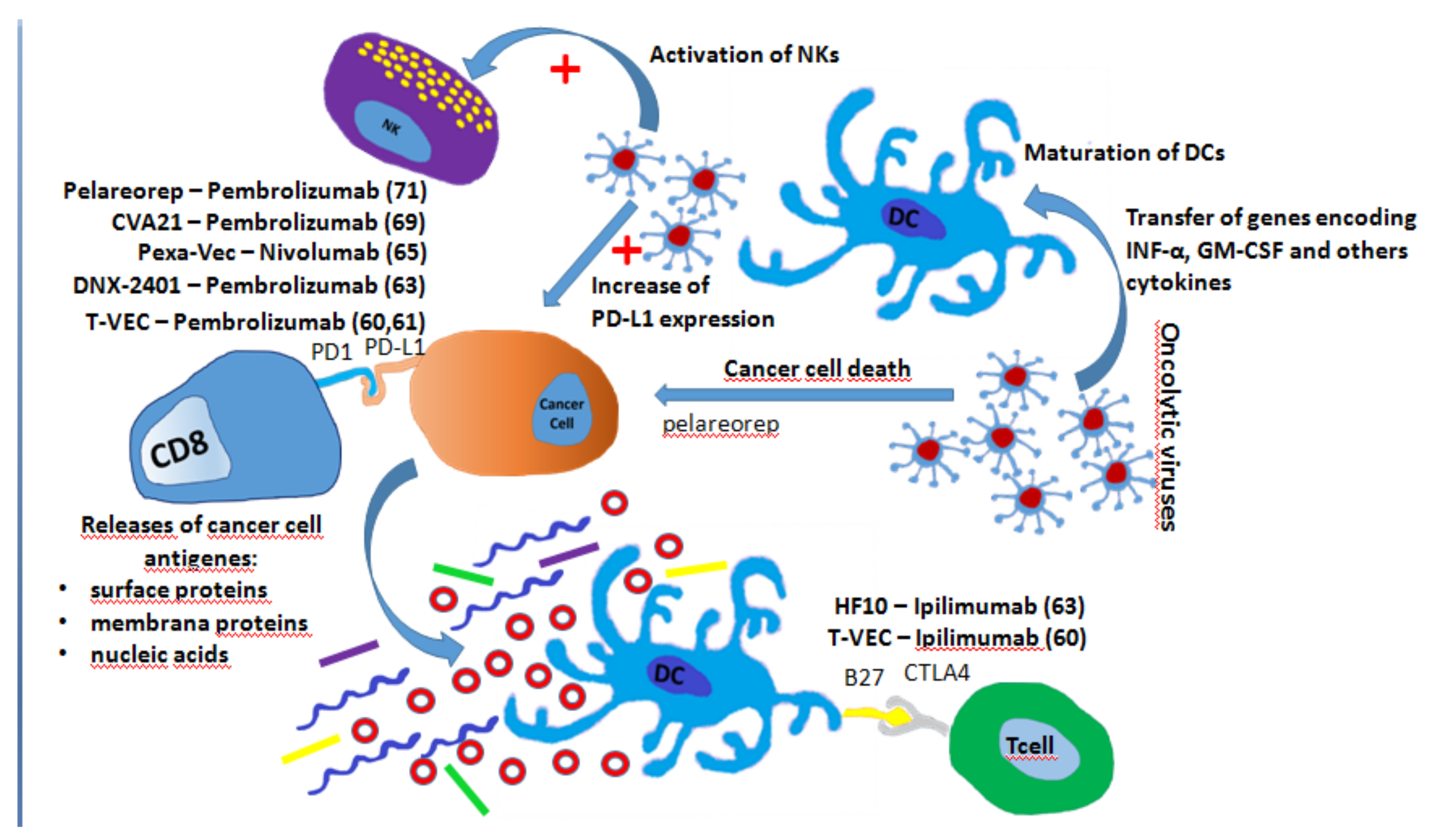
7. Oncolytic Virus and ICIs
8. Small Molecule Inhibitors and ICIs
9. Conclusions
Funding
Conflicts of Interest
References
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Cella, D.; Grünwald, V.; Escudier, B.; Hammers, H.J.; George, S.; Nathan, P.; Grimm, M.O.; Rini, B.I.; Doan, J.; Ivanescu, C.; et al. Patient-reported outcomes of patients with advancedrenalcell carcinoma treated withnivolumabplusipilimumabversussunitinib (CheckMate214): A randomised, phase 3 trial. Lancet Oncol. 2019, 20, 297–310. [Google Scholar] [CrossRef]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-linepembrolizumabin cisplatin-ineligible patients with locally advanced and unresectable or metastaticurothelialcancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Kanjanapan, Y.; Day, D.; Wang, L.; Al-Sawaihey, H.; Abbas, E.; Namini, A.; Siu, L.L.; Hansen, A.; Razak, A.A.; Spreafico, A.; et al. Hyperprogressive disease in early-phase immunotherapy trials: Clinical predictors and association with immune-related toxicities. Cancer 2019, 125, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Chuong, M.; Chang, E.T.; Choi, E.Y.; Mahmood, J.; Lapidus, R.G.; Davila, E.; Carrier, F. Exploring the Concept of Radiation “Booster Shot” in Combination with an Anti-PD-L1 mAb to Enhance Anti-Tumor Immune Effects in Mouse Pancreas Tumors. J. Clin. Oncol. Res. 2017, 5, 1058. [Google Scholar]
- Meng, X.; Feng, R.; Yang, L.; Xing, L.; Yu, J. The Role of Radiation Oncology in Immuno-Oncology. Oncologist 2019, 24, S42–S52. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Honda, K. Intestinal commensal microbes as immune modulators. Cell. Host Microbe. 2012, 12, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Roy, S.; Trinchieri, G. Microbiota: A key orchestrator of cancer therapy. Nat. Rev. Cancer 2017, 5, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Gomperts, B.I.; Lepage, P.; Chamaillard, M.; Zitvogel, L. Enhancing the clinical coverage and anticancer efficacy of immune checkpoint blockade through manipulation of the gut microbiota. Oncoimmunology 2016, 6, e1132137. [Google Scholar] [CrossRef] [PubMed]
- Dubin, K.; Callahan, M.K.; Ren, B.; Khanin, R.; Viale, A.; Ling, L.; No, D.; Gobourne, A.; Littmann, E.; Huttenhower, C.; et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. 2016, 7, e10391. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Baseline gut micro biota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 2017, 28, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 5, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Shen, L.; Wang, Y.; Liu, Q.; Goodwin, T.J.; Li, J.; Dorosheva, O.; Liu, T.; Liu, R.; Huang, L. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun. 2018, 9, 2237. [Google Scholar] [CrossRef]
- Mkrtichyan, M.; Najjar, Y.G.; Raulfs, E.C.; Abdalla, M.Y.; Samara, R.; Rotem-Yehudar, R.; Cook, L.; Khleif, S.N. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur. J. Immunol. 2011, 41, 2977–2986. [Google Scholar] [CrossRef]
- Merlano, M.C.; Merlotti, A.M.; Licitra, L.; Denaro, N.; Fea, E.; Galizia, D.; Di Maio, M.; Fruttero, C.; Curcio, P.; Vecchio, S.; et al. Activation of immune responses in patients with relapsed-metastatic head and neck cancer (CONFRONT phase I-II trial): Multimodality immunotherapy with avelumab, short-course radiotherapy, and cyclophosphamide. Clin. Transl. Radiat. Oncol. 2018, 12, 47–52. [Google Scholar] [CrossRef]
- Cui, S. Immunogenic Chemotherapy Sensitizes Renal Cancer to Immune Checkpoint Blockade Therapy in Preclinical Models. Med. Sci. Monit. 2017, 23, 3360–3366. [Google Scholar] [CrossRef]
- VanDer Kraak, L.; Goel, G.; Ramanan, K.; Kaltenmeier, C.; Zhang, L.; Normolle, D.P.; Freeman, G.J.; Tang, D.; Nason, K.S.; Davison, J.M.; et al. 5-Fluorouracil upregulates cell surface B7-H1 (PD-L1) expression in gastrointestinal cancers. J. Immunother. Cancer 2016, 4, 65. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, H.; Meng, F.; Li, J.; Zhang, S. Combined Trabectedin and anti-PD1 antibody produces a synergistic antitumor effect in a murine modelofovarian cancer. J. Transl. Med. 2015, 13, 247. [Google Scholar] [CrossRef]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-κB to Foster an Immunosuppressive Tumor Microenvironment in OvarianCancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef]
- Mesnage, S.J.L.; Auguste, A.; Genestie, C.; Dunant, A.; Pain, E.; Drusch, F.; Gouy, S.; Morice, P.; Bentivegna, E.; Lhomme, C.; et al. Neoadjuvant chemotherapy (NACT) increases immune infiltration and programmed death-ligand 1 (PD-L1) expression in epithelialovariancancer (EOC). Ann. Oncol. 2017, 28, 651–657. [Google Scholar] [CrossRef]
- Kok, M.; Voorwerk, L.; Horlings, H.; Sikorska, K.; van der Vijver, K.; Slagter, M.; Warren, S.; Ong, S.; Wiersma, T.; Russell, N.; et al. Adaptive phase II randomized trial of nivolumab after induction treatment in triple negative breast cancer (TONIC trial): Final response data stage I and first translational data. J. Clin. Oncol. 2018, 36, 1012. [Google Scholar] [CrossRef]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.B.; Blohmer, J.U.; Grischke, E.M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; Rezai, M.; et al. Randomized phase II neoadjuvant study (GeparNuevo) to investigate the addition of durvalumab to a taxane-anthracycline containing chemotherapy in triple negative breast cancer (TNBC). J. Clin. Oncol. 2018, 36, 104. [Google Scholar] [CrossRef]
- NCT03585465: Nivolumab in Combination with Metronomic Chemotherapy in Paediatrics Refractory/Relapsing Solid Tumors or Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03585465 (accessed on 30 October 2018).
- NCT03683407: Effect of Chemotherapy on TMB in NSCLC. Available online: https://clinicaltrials.gov/ct2/show/NCT03683407 (accessed on 30 October 2018).
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Stockmann, C.; Schadendorf, D.; Klose, R.; Helfrich, I. The impact of the immune system on tumor: Angiogenesis and vascular remodeling. Front. Oncol. 2014, 4, e69. [Google Scholar] [CrossRef] [PubMed]
- Nuti, M.; Zizzari, I.G.; Botticelli, A.; Rughetti, A.; Marchetti, P. The ambitious role of anti angiogenesis molecules: Turning a cold tumor into a hot one. Cancer Treat. Rev. 2018, 70, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Matsumoto, K.; Takahashi, M.; Mukohara, T.; Futamura, M.; Masuda, N.; Tsurutani, J.; Yoshimura, K.; Minami, H.; Takano, T. Phase II study of a combination therapy of nivolumab, bevacizumab and paclitaxel in patients with HER2-negative metastatic breast cancer as a first-line treatment (WJOG9917B, NEWBEAT trial). J. Clin. Oncol. 2018, 36, TPS1110. [Google Scholar] [CrossRef]
- Liu, J.F.; Herold, C.; Luo, W.; Penson, R.; Horowitz, N.; Konstantinopoulos, P.; Castro, C.; Curtis, J.; Matulonis, U.A.; Cannistra, S.; et al. A phase 2 trial of combination nivolumab and bevacizumab in recurrent ovarian cancer. Ann. Oncol. 2018, 29, viii332–viii358. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Antonia, S.J.; Shepherd, F.A.; Chow, L.Q.; Goldman, J.; Shen, Y.; Chen, A.C.; Gettinger, S. Nivolumab (Anti-PD-1; BMS-936558, ONO-4538) Maintenance as Monotherapy or in Combination With Bevacizumab (BEV) for Non-Small Cell Lung Cancer (NSCLC) Previously Treated With Chemotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, S32. [Google Scholar] [CrossRef]
- Motzer, R.J.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. IMmotion151: A Randomized Phase III Study of Atezolizumab Plus Bevacizumab vs. Sunitinib in Untreated Metastatic Renal Cell Carcinoma (mRCC). J. Clin. Oncol. 2018, 36, 578. [Google Scholar] [CrossRef]
- NCT024243324: A Study of Ramucirumab Plus Pembrolizumab in Participants with Gastric or GEJ Adenocarcinoma, NSCLC, Transitional Cell Carcinoma of the Urothelium, or Biliary Tract Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02443324 (accessed on 30 October 2018).
- Chau, I.; Penel, N.; Arkenau, H.T.; Santana-Davila, R.; Calvo, E.; Soriano, A.O.; Mi, G.; Jin, J.; Ferry, D.; Herbst, R.S.; et al. Safety and antitumor activity of ramucirumab plus pembrolizumab in treatment naïve advanced gastric or gastroesophageal junction (G/GEJ) adenocarcinoma: Preliminary results from a multi-disease phase I study (JVDF). J. Clin. Oncol. 2018, 36, 101. [Google Scholar] [CrossRef]
- Amin, A.; Plimack, E.R.; Ernstoff, M.S.; Lewis, L.D.; Bauer, T.M.; McDermott, D.F.; Carducci, M.; Kollmannsberger, C.; Rini, B.I.; Heng, D.Y.C.; et al. Safety and efficacy of nivolumab in combination with sunitinib or pazopanib in advanced or metastatic renal cell carcinoma: The CheckMate 016 study. J. Immunother. Cancer 2018, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapyand new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGITand PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef] [PubMed]
- NCT03563716: A Study of MTIG7192A in Combination withAtezolizumab in Chemotherapy-Naïve Patients with Locally Advanced or Metastatic Non-Small Cell Lung Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03563716 (accessed on 30 October 2018).
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Perez Gracia, J.L.; Haanen, J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Bono, P.; Bhatia, S.; Melero, I.; Nyakas, M.S.; Svane, I.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S.; et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J. Clin. Oncol. 2017, 35, 9520. [Google Scholar] [CrossRef]
- Hong, D.S.; Schoffski, P.; Calvo, A.; Sarantopoulos, J.; De Olza, M.O.; Carvajal, R.D.; Prawira, A.; Kyi, C.; Esaki, T.; Akerley, W.L.; et al. Phase I/II study of LAG525 ± spartalizumab (PDR001) in patients (pts) with advanced malignancies. J. Clin. Oncol. 2018, 36, 3012. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- NCT02817633: A Phase 1 Study of TSR-022, an Anti-TIM-3 Monoclonal Antibody, in Patients with Advanced Solid Tumors (AMBER). Available online: https://clinicaltrials.gov/ct2/show/NCT02817633 (accessed on 30 October 2018).
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, C.J.; Warner, S.G. Oncolytic viruses and checkpoint inhibitors: Combination therapy in clinical trials. Clin. Transl. Med. 2018, 7, 35. [Google Scholar] [CrossRef]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of TalimogeneLaherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef]
- NCT02263508: Pembrolizumab with or without TalimogeneLaherparepvec or TalimogeneLaherparepvec Placebo in Unresected Melanoma (KEYNOTE-034). Available online: https://clinicaltrials.gov/ct2/show/NCT02263508 (accessed on 30 October 2018).
- Andtbacka, R.H.I.; Ross, M.I.; Agarwala, S.S.; Taylor, M.H.; Vetto, J.T.; Neves, R.I.; Daud, A.; Khong, H.T.; Ungerleider, R.S.; Tanaka, M.; et al. Final results of a phase II multicenter trial of HF10, a replication-competent HSV-1 oncolytic virus, and ipilimumab combination treatment in patients with stage IIIB-IV unresectable or metastatic melanoma. J. Clin. Oncol. 2017, 35, 9510. [Google Scholar] [CrossRef]
- NCT02798406: Combination Adenovirus + Pembrolizumab to Trigger Immune Virus Effects (CAPTIVE). Available online: https://clinicaltrials.gov/ct2/show/NCT02798406 (accessed on 30 October 2018).
- Haddad, D. Genetically Engineered Vaccinia Viruses As Agents for Cancer Treatment, Imaging, and Transgene Delivery. Front. Oncol. 2017, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Anthoney, A.; Samson, A.; West, E.; Turnbull, S.J.; Scott, K.; Tidswell, E.; Kingston, J.; Johnpulle, M.; Noutch, S.; Bendjama, K.; et al. Single intravenous preoperative administration of the oncolytic virus Pexa-Vec to prime anti-tumor immunity. J. Clin. Oncol. 2018, 36, 3092. [Google Scholar] [CrossRef]
- NCT03206073: A Phase I/II Study of Pexa-Vec Oncolytic Virus in Combination with Immune Checkpoint Inhibition in Refractory Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03206073 (accessed on 30 October 2018).
- NCT02977156: Immunization Strategy with Intra-tumoral Injections of Pexa-Vec With Ipilimumab in Metastatic/Advanced Solid Tumors. (ISI-JX). Available online: https://clinicaltrials.gov/ct2/show/NCT02977156 (accessed on 30 October 2018).
- NCT02307149: Intratumoral CAVATAK (CVA21) and Ipilimumab in Patients with Advanced Melanoma (VLA-013 MITCI) (MITCI). Available online: https://clinicaltrials.gov/ct2/show/NCT02307149 (accessed on 30 October 2018).
- Silk, A.W.; Kaufman, H.; Gabrail, N.; Mehnert, J.; Bryan, J.; Norrell, J.; Medina, D.; Bommareddy, P.; Shafren, D.; Grose, M.; et al. Phase 1b study of intratumoralCoxsackievirus A21 (CVA 21) and systemic pembrolizumab in a dvanced melanoma patients: Interim results of the CAPRA clinical trial. Cancer Res. 2017, 77, CT026. [Google Scholar] [CrossRef]
- Mahalingam, D.; Fountzilas, C.; Moseley, J.L.; Noronha, N.; Cheetham, K.; Dzugalo, A.; Nuovo, G.; Gutierrez, A.; Arora, S.P.; et al. A study of REOLYSIN in combination with pembrolizumab and chemotherapy in patients (pts) with relapsed metastatic adenocarcinoma of the pancreas (MAP). J. Clin. Oncol. 2017, 35, e15753. [Google Scholar] [CrossRef]
- NCT02620423: Study of Pembrolizumab with REOLYSIN® and Chemotherapy in Patients with Advanced Pancreatic Adenocarcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02620423 (accessed on 30 October 2018).
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef]
- Comin-Anduix, B.; Chodon, T.; Sazegar, H.; Matsunaga, D.; Mock, S.; Jalil, J.; Escuin-Ordinas, H.; Chmielowski, B.; Koya, R.C.; Ribas, A.; et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin. Cancer Res. 2010, 16, 6040–6048. [Google Scholar] [CrossRef] [PubMed]
- Wilmott, J.S.; Long, G.V.; Howle, J.R.; Haydu, L.E.; Sharma, R.N.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Scolyer, R.A. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin. Cancer Res. 2012, 18, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.D.; Chen, Z.; Melendez, B.; Talukder, A.; Khalili, J.S.; Rodriguez-Cruz, T.; Liu, S.; Whittington, M.; Deng, W.; Li, F.; et al. BRAFV600E co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol. Res. 2015, 3, 602–609. [Google Scholar] [CrossRef]
- Koya, R.C.; Mok, S.; Otte, N.; Blacketor, K.J.; Comin-Anduix, B.; Tumeh, P.C.; Minasyan, A.; Graham, N.A.; Graeber, T.G.; Chodon, T.; et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012, 72, 3928–3937. [Google Scholar] [CrossRef]
- Knight, D.A.; Ngiow, S.F.; Li, M.; Parmenter, T.; Mok, S.; Cass, A.; Haynes, N.M.; Kinross, K.; Yagita, H.; Koya, R.C.; et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J. Clin. Investig. 2016, 126, 402–403. [Google Scholar] [CrossRef]
- Hooijkaas, A.; Gadiot, J.; Morrow, M.; Stewart, R.; Schumacher, T.; Blank, C.U. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology 2012, 1, 609–617. [Google Scholar] [CrossRef]
- Cooper, Z.A.; Juneja, V.R.; Sage, P.T.; Frederick, D.T.; Piris, A.; Mitra, D.; Lo, J.A.; Hodi, F.S.; Freeman, G.J.; Bosenberg, M.W.; et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol. Res. 2014, 2, 643–654. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Moreno, B.H.; Tsoi, J.; Faja, L.R.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef]
- Ribas, A.; Hodi, F.S.; Callahan, M.; Konto, C.; Wolchok, J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med. 2013, 368, 1365–1366. [Google Scholar] [CrossRef]
- Minor, D.R.; Puzanov, I.; Callahan, M.K.; Hug, B.A.; Hoos, A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res. 2015, 28, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Callahan, M.; Gerald, P.; Linette, G.; Luke, J.J.; Sosmanet, J.A.; Wolchok, J.D.; Hamid, O.; Minor, D.R.; Orford, K.W.; et al. Phase 1 study of the BRAF inhibitor dabrafenib (D) with or without the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation–positive unresectable or metastatic melanoma (MM). J. Clin. Oncol. 2014, 32, 2511. [Google Scholar] [CrossRef]
- NCT01656642: A Phase 1b Study of Atezolizumab in Combination with BRAFV600-Mutation Positive Metastatic Melanoma. Available online: https://clinicaltrials.gov/ct2/show/NCT01656642 (accessed on 30 October 2018).
- Ribas, A.; Hodi, F.; Lawrence, D.; Atkinson, V.; Starodub, A.; Carlino, M.S.; Fisher, R.A.; Long, G.V.; Miller, W.H.; Huang, Y.; et al. Pembrolizumab (pembro) in combination with dabrafenib (D) and trametinib (T) for BRAF-mutant advanced melanoma: Phase 1 KEYNOTE-022 study. J. Clin. Oncol. 2016, 34, 3014. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Gonzalez, R.; Lewis, K.D.; Hamid, O.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Wallin, J.; Pitcher, B.; Cha, E.; et al. Atezolizumab (A) + cobimetinib (C) + vemurafenib (V) in BRAFV600-mutant metastatic melanoma (mel): Updated safety and clinical activity. J. Clin. Oncol. 2017, 35, 3063. [Google Scholar] [CrossRef]
- Larmonier, N.; Janikashvili, N.; La Casse, C.J.; Larmonier, C.B.; Cantrell, J.; Situ, E.; Lundeen, T.; Bonnotte, B.; Katsanis, E. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL-tumors. J. Immunol. 2008, 181, 6955–6963. [Google Scholar] [CrossRef] [PubMed]
- Ménard, C.; Blay, J.Y.; Borg, C.; Michiels, S.; Ghiringhelli, F.; Robert, C.; Nonn, C.; Chaput, N.; Taïeb, J.; Delahaye, N.F.; et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. 2009, 69, 3563–3569. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.M.; Zeng, S.; Zhang, J.Q.; Kim, T.S.; Cohen, N.A.; Beckman, M.J.; Medina, B.D.; Maltbaek, J.H.; Loo, J.K.; Crawley, M.H.; et al. PD-1/PD-L1 Blockade Enhances T-cell Activity and Antitumor Efficacy of Imatinib in Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2017, 23, 454–465. [Google Scholar] [CrossRef]
- NCT01738139: Ipilimumab and Imatinib Mesylate in Treating Participants with Metastatic or Unresectable Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT01738139 (accessed on 30 October 2018).
- Reilley, M.J.; Bailey, A.; Subbiah, V.; Janku, F.; Naing, A.; Falchook, G.; Karp, D.; Piha-Paul, S.; Tsimberidou, A.; Fu, S.; et al. Phase I clinical trial of combination imatinib and ipilimumab in patients with advanced malignancies. J. Immunother. Cancer 2017, 5, 35. [Google Scholar] [CrossRef]
- Cash, H.; Shay, S.; Moree, E.; Cariso, A.; Uppaluri, R.; Van Waes, C.; Allen, C. mTOR and MEK1/2 inhibition differentially modulate tumor growth and the immune microenvironment in syngeneic models of oral cavity cancer. Onco. Target 2015, 6, 36400–36417. [Google Scholar] [CrossRef]
- Moore, E.C.; Cash, H.A.; Caruso, A.M.; Uppaluri, R.; Hodge, J.W.; Van Waes, C.; Allen, C.T. Enhanced tumor control with combination mTOR and PD-L1 inhibition in syngeneic oral cavity cancers. Cancer Immunol. Res. 2016, 4, 611–620. [Google Scholar] [CrossRef]
- Goel, S.; De Cristo, M.J.; Watt, A.C.; Brin Jones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Teo, Z.L.; Versaci, S.; Dushyanthen, S.; Caramia, F.; Savas, P.; Mintoff, C.P.; Zethoven, M.; Virassamy, B.; Luen, S.J.; McArthur, G.A.; et al. Combined CDK4/6 and PI3Kα Inhibition Is Synergistic and Immunogenic in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 6340–6352. [Google Scholar] [CrossRef]
- NCT02130466: A Study of the Safety and Efficacy of Pembrolizumab (MK-3475) in Combination with Trametinib and Dabrafenib in Participants with Advanced Melanoma (MK-3475-022/KEYNOTE-022). Available online: https://clinicaltrials.gov/ct2/show/NCT02130466 (accessed on 30 October 2018).
- Nanda, V.G.Y.; Peng, W.; Hwu, P.; Davies, M.A.; Ciliberto, G.; Fattore, L.; Malpicci, D.; Aurisicchio, L.; Ascierto, P.A.; Croce, C.M.; et al. Melanoma and immunotherapy bridge 2015. Naples, Italy. 1–5 December 2015. J. Transl. Med. 2016, 14 (Suppl. 1), 65. [Google Scholar] [CrossRef] [PubMed]
- Borg, C.; Terme, M.; Taïeb, J.; Ménard, C.; Flament, C.; Robert, C.; Maruyama, K.; Wakasugi, H.; Angevin, E.; Thielemans, K.; et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J. Clin.Investig. 2004, 114, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, M.; Savage, N.D.; Jordanova, E.S.; Briaire-de Bruijn, I.H.; Walburg, K.V.; Ottenhoff, T.H.; Hogendoorn, P.C.; van der Burg, S.H.; Gelderblom, H.; van Hall, T. Anti-inflammatory M2 type macrophages characterize metastasized and tyrosine kinase inhibitor-treated gastrointestinal stromal tumors. Int. J. Cancer 2010, 127, 899–909. [Google Scholar] [CrossRef]
- Loukinova, E.; Dong, G.; Enamorado-Ayalya, I.; Thomas, G.R.; Chen, Z.; Schreiber, H.; VanWaes, C. Growth regulated oncogene-alpha expression by murine squamous cell carcinoma promotes tumor growth, metastasis, leukocyte infiltration and angiogenesis by a host CXC receptor-2 dependent mechanism. Oncogene 2000, 19, 3477–3486. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, J.J.; Salem, J.E.; Sosman, J.A.; Lebrun-Vignes, B.; Johnson, D.B. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet 2018, 391, 933. [Google Scholar] [CrossRef]
- Tajiri, K.; Ieda, M. Cardiac Complications in Immune Checkpoint Inhibition Therapy. Front. Cardiovasc. Med. 2019, 6, 3. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Immune Therapy | Enhancer |
|---|---|
| ‘immune checkpoint inhibitors’, ‘anti-PD-(L)1’, ‘anti-CTLA-4’. | “microbiote” OR “microbiota” OR “gut microbe” OR “bacteria” |
| ‘immune checkpoint inhibitors’, ‘anti-PD-(L)1’, ‘anti-CTLA-4’. | “chemotherapy” OR“ chemotherapeutics” OR “metronomic chemotherapy” |
| ‘immune checkpoint inhibitors’, ‘anti-PD-(L)1’, ‘anti-CTLA-4’. | “anti-angiogenetic therapies” OR “bevacizumab” OR “nintedanib” OR “Aflibercept” OR “pazopanib” OR “sunitinib” |
| ‘immune checkpoint inhibitors’, ‘anti-PD-(L)1’, ‘anti-CTLA-4’. | “co-inhibitor receptors” OR “TIGIT” OR “LAG3” OR “TIM-3” |
| ‘immune checkpoint inhibitors’, ‘anti-PD-(L)1’, ‘anti-CTLA-4’. | “Oncolytic virus” OR “adenovirus” OR ”vaccinia viruses” OR ”Coxsackieviruses” OR ”Reoviruses” |
| ‘immune checkpoint inhibitors’, ‘anti-PD-(L)1’, ‘anti-CTLA-4’. | “small molecules” OR “tyrosine kinase inhibitor” OR “mTOR inhibitor” OR “cyclin inhibitor” |
| Small Molecule Enhancer | ICI | Cancer | Study Design | Results/Enhancing | Reference |
|---|---|---|---|---|---|
| BRAFi | Not associated | Melanoma | In vitro | BRAF inhibition enhance melanoma antigen expression | Wilmott, 2013. [72] |
| Selective BRAF inhibitors | Not associated | Melanoma | In vitro | Induction of Tcell infiltration into human metastatic melanoma Up-regulation of PD-L1 in tumor microenvironment | Wilmott, 2012. [74] |
| Dabrafenib and trametinib | (pmel-1 adoptive cell transfer) | BRAFV600E driven melanoma | In vivo—mouse model | Complete tumor regression with increased T cell infiltration into tumors and improved in vivo cytotoxicity | Cooper, 2014. [79] |
| Dabrafenib and trametinib | anti-PD1 | SM1 tumors (melanoma) | In vivo—mouse model | Superior anti-tumor effect compared to the results obtained with the only small molecules combination | Hu-Lieskovan, 2015. [80] |
| Vemurafenib | Ipilimumab | Melanoma | Phase 1 trial | Stopped after one month due to liver toxicity | Ribas, 2013. [81] |
| Dabrafenib, trametinib | Ipilimumab | Melanoma | Phase 1 trial | Stopped due to excessive colon toxicity | Minor, 2015. [82] |
| Dabrafenib | Ipilimumab | BRAF-mutated melanoma | Phase 1 trial | ORR of 69% Good safety profile | Puzanov, 2014. [83] |
| Dabrafenib and trametinib | pembrolizumab | BRAF-mutated melanoma | KEYNOTE-022, an ongoing phase I/II trial | ORR of 60% (n = 9 PR, n = 2 SD, n = 3 PD) | NCT02130466, [84] Ribas, 2016. [85] |
| Vemurafenib (V) | Atezolizumab (A) | Melanoma | Phase Ib trial (V-run in vs. concurrent V-A) | Higher ORR was seen with V run-in than with concurrent A + V start | Sullivan, 2016. [86] |
| Vemurafenib, and cobimetinib | Atezolizumab | BRAFV600-mutant melanoma | Phase I/II trial | Manageable safety profile and promising antitumor activity | NCT01656642. [84] |
| imatinib | Not associated | GIST | In vitro study | Reduction of Treg immunosuppressive function | Larmonier, 2008. [87] |
| Imatinib | Not associated | GIST | In vivo study | PFS correlated with IFN-γ secretion by NK cells | Ménard, 2009. [88] |
| Imatinib | Not associated | GIST | In vivo—mouse model | Activated CD8+ T cells and induced Treg apoptosis in tumor sample | Balachandran, 2011. [89] |
| Imatinib | Anti-PD-1 (RMP1-14) or anti-PD-L1 (10F.9G2) | GIST | In vivo—mouse model | Increased antitumor effects by enhancing cytotoxic T cell effector function | Seifert, 2017. [90] |
| Imatinib | Ipilimumab | GIST and other c-Kit positive solid cancers | Phase 1 trial | Manageable safety profile in multiple tumor types. Low activity with no clear signal for synergy in escalation or GIST expansion cohorts | NCT01738139, [91] Reilley, 2017. [92] |
| Rapamycin | Not associated | Oral cancer | In vivo—mouse model | Reduction of tumor growth through CD8-activity | Cash, 2015. [93] |
| Rapamycin | Not associated | Oral cancer | In vivo—mouse model | Enhancing of IFNγ production by peripheral and tumor-infiltrating CD8 T cells | Moore, 2016. [94] |
| Rapamycin | PD-L1 mAb | Oral cancer | In vivo—mouse model | Activation of CD8 T cells in tumor infiltration increased by the addition of rapamycin | Moore, 2016. [94] |
| CDK4/6 inhibitor | Not associated | Breast cancer | In vivo—mouse model/blood sample patients | Anti-tumor immunity through proliferation of Tregs | Goel, 2017. [95] |
| CDK4/6 inhibitor | anti-PD-1 | Breast cancer | In vivo—mouse model | Enhancing of tumor regression and dramatically improving of OS | Zhang, 2018. [96] |
| CDK4/6 inhibitor and PI3K antagonist | Anti PD-1 and anti CTLA-4 | Triple negative breast cancer | In vivo—mouse model | Inhibition induced complete and durable regressions (> one year) of breast tumors in in vivo models. | Teo, 2017. [97] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longo, V.; Brunetti, O.; Azzariti, A.; Galetta, D.; Nardulli, P.; Leonetti, F.; Silvestris, N. Strategies to Improve Cancer Immune Checkpoint Inhibitors Efficacy, Other Than Abscopal Effect: A Systematic Review. Cancers 2019, 11, 539. https://doi.org/10.3390/cancers11040539
Longo V, Brunetti O, Azzariti A, Galetta D, Nardulli P, Leonetti F, Silvestris N. Strategies to Improve Cancer Immune Checkpoint Inhibitors Efficacy, Other Than Abscopal Effect: A Systematic Review. Cancers. 2019; 11(4):539. https://doi.org/10.3390/cancers11040539
Chicago/Turabian StyleLongo, Vito, Oronzo Brunetti, Amalia Azzariti, Domenico Galetta, Patrizia Nardulli, Francesco Leonetti, and Nicola Silvestris. 2019. "Strategies to Improve Cancer Immune Checkpoint Inhibitors Efficacy, Other Than Abscopal Effect: A Systematic Review" Cancers 11, no. 4: 539. https://doi.org/10.3390/cancers11040539
APA StyleLongo, V., Brunetti, O., Azzariti, A., Galetta, D., Nardulli, P., Leonetti, F., & Silvestris, N. (2019). Strategies to Improve Cancer Immune Checkpoint Inhibitors Efficacy, Other Than Abscopal Effect: A Systematic Review. Cancers, 11(4), 539. https://doi.org/10.3390/cancers11040539