Efficacy of a Selective Binder of αVβ3 Integrin Linked to the Tyrosine Kinase Inhibitor Sunitinib in Ovarian Carcinoma Preclinical Models

 , , and
, , and 
Abstract
1. Introduction
2. Results
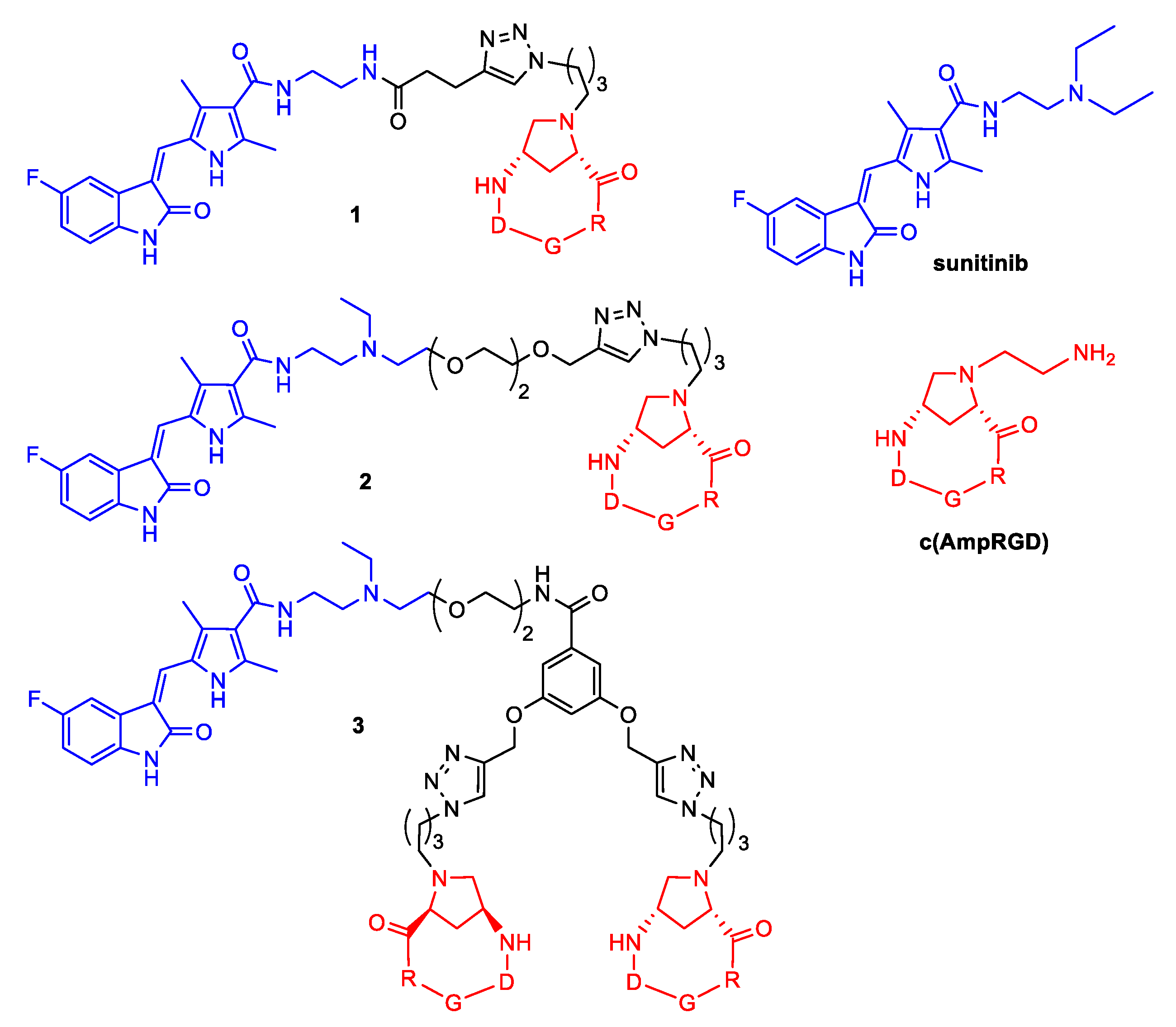
2.1. Synthesis of Conjugates 1–3
2.2. Levels of VEGFR2
2.3. Cell Sensitivity of Cisplatin-Sensitive and -Resistant IGROV-1 Ovarian Carcinoma Cell Lines
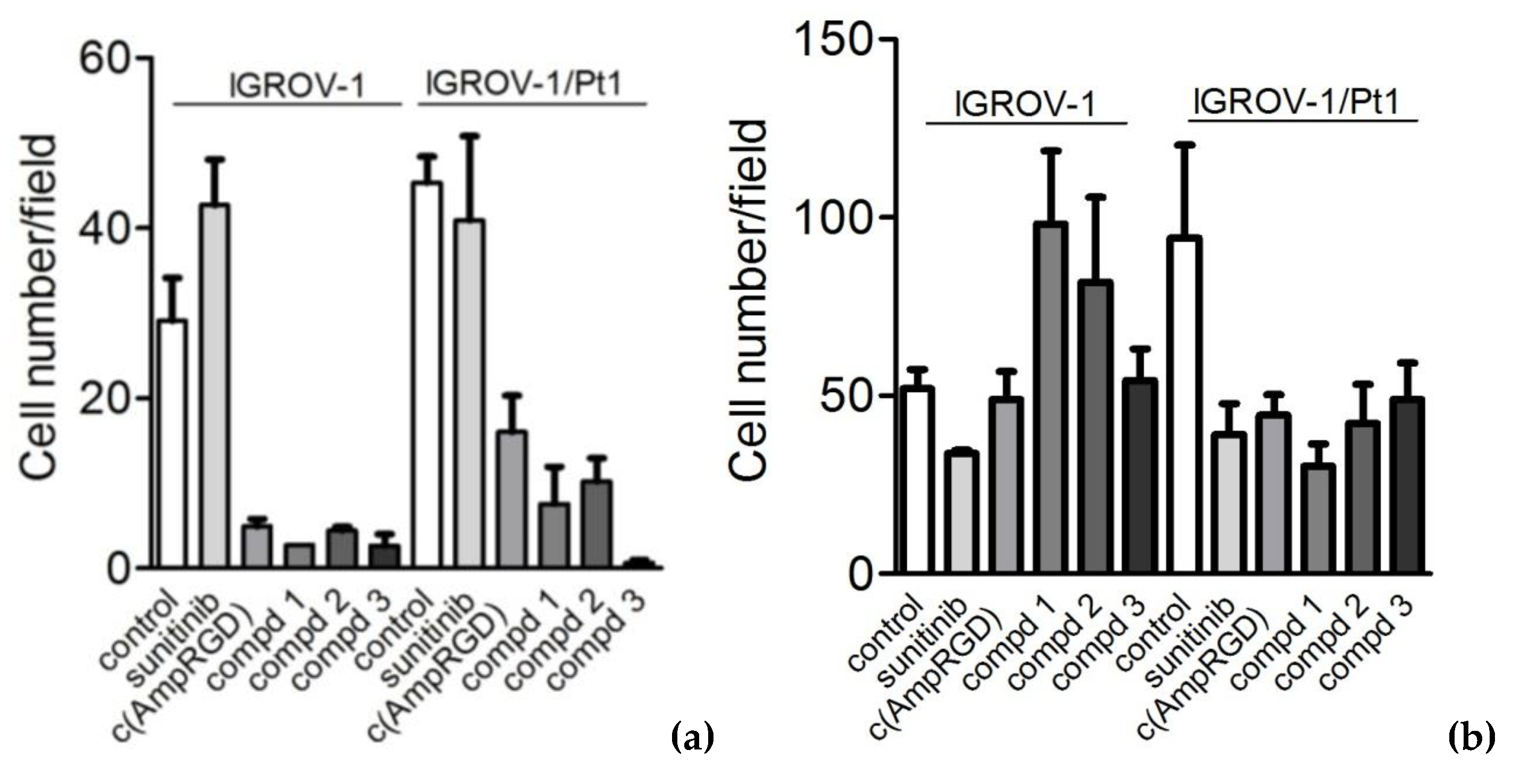
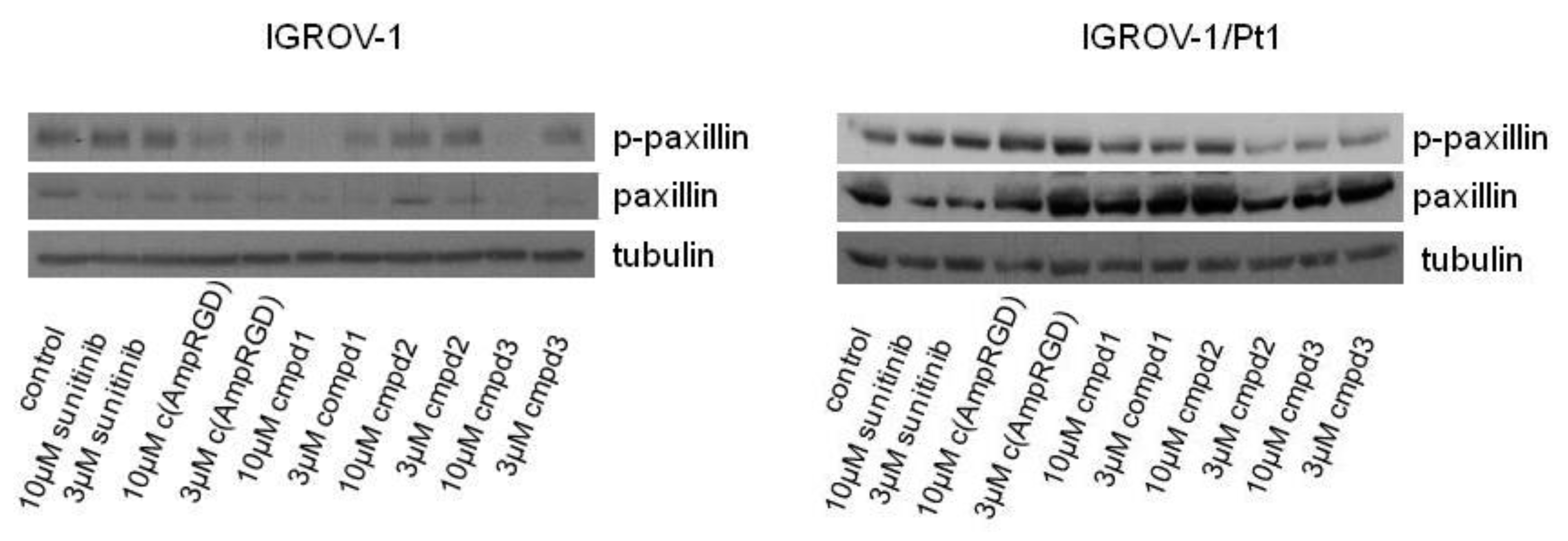
2.4. Modulation of Cell Migratory and Invasive Abilities and Modulation of Downstream Targets of VEGFR2 of Cisplatin-Sensitive and -Resistant IGROV-1 Ovarian Carcinoma Cell Lines
2.5. Cell Sensitivity and Modulation of Cell Migratory and Invasive Abilities of Cisplatin-Sensitive A2780 Ovarian Carcinoma Cell Line
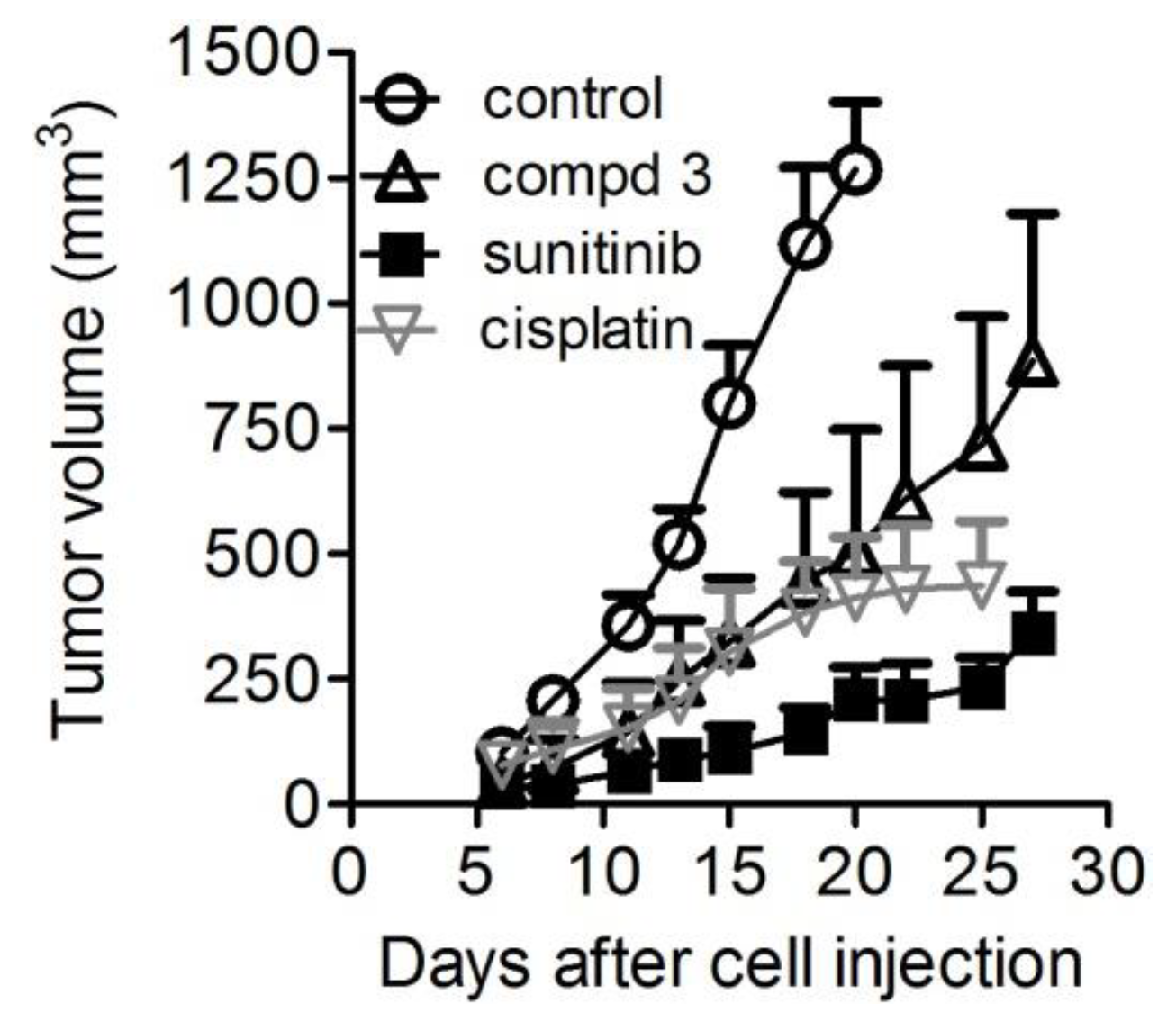
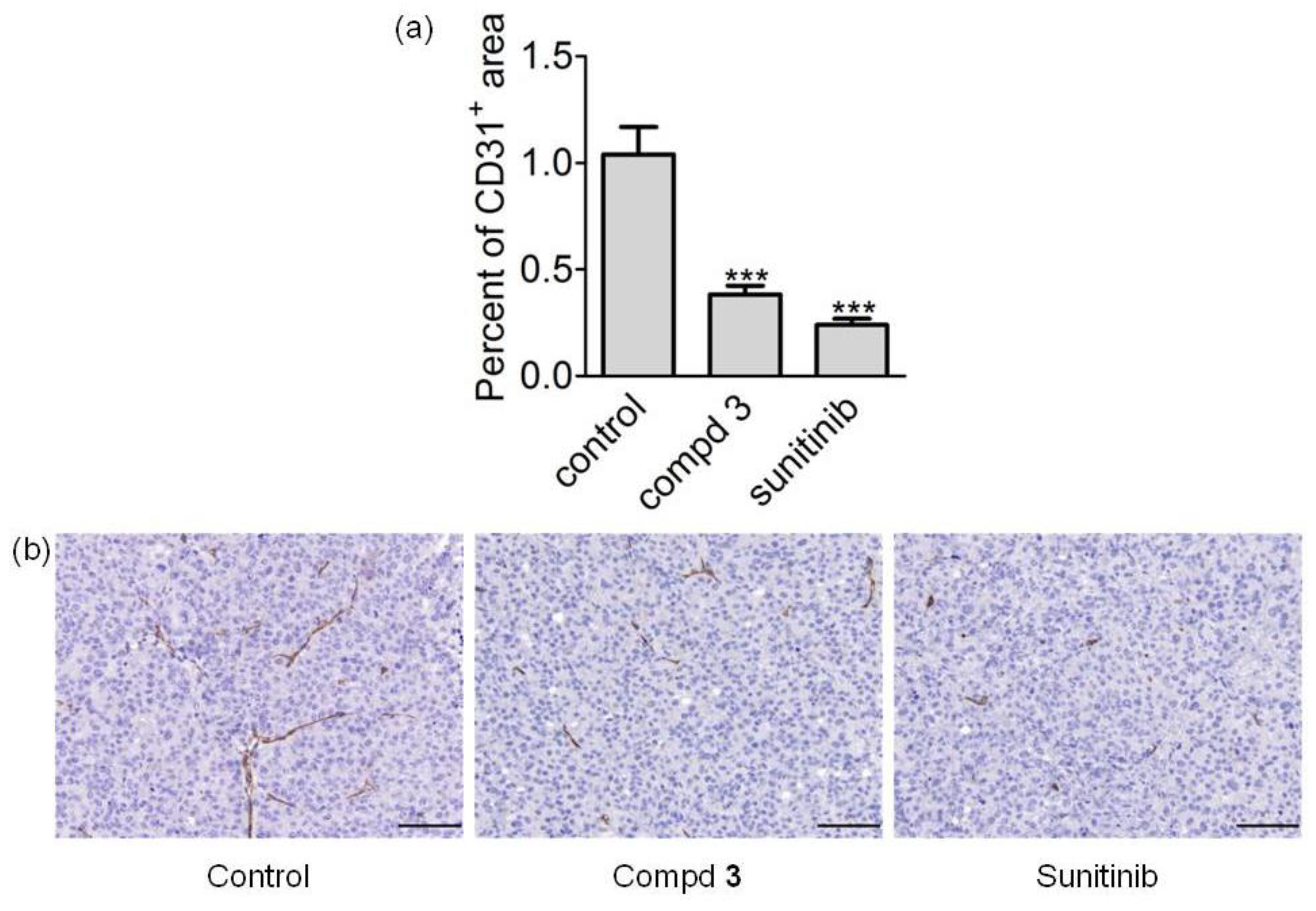
2.6. In Vivo Antitumor Activity
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Lines and Growth Conditions
4.3. Cell Migration and Invasion Assays
4.4. Western Blot Analyses
4.5. Analysis of Integrin Levels
4.6. Antitumor Activity Evaluation
4.7. Immunohistochemistry and Digital Image Analysis
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer. J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of Ovarian cancer: New opportunities for translation. Nat. Rev. Cancer. 2009, 9, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Cannistra, S.A. Cancer of the ovary. N. Engl. J. Med. 2004, 351, 2519–2529. [Google Scholar] [CrossRef]
- Kelland, L. The Resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer. 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Cossa, G.; Gatti, L.; Zunino, F.; Perego, P. Strategies to improve the efficacy of platinum compounds. Curr. Med. Chem. 2009, 16, 2355–2365. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Zuco, V.; Gatti, L.; Lanzi, C.; Zaffaroni, N.; Colombo, D.; Perego, P. Targeting the Akt kinase to modulate survival, invasiveness and drug resistance of cancer cells. Curr. Med. Chem. 2013, 20, 1923–1945. [Google Scholar] [CrossRef]
- Ouyang, W.; Xu, L.; Huang, Z.; Guo, J.; Cai, J.; Gao, X.; Wang, Z. Role of HER family members in predicting.prognoses in epithelial ovarian cancer: A meta-analysis. Tumori 2015, 101, 595–602. [Google Scholar] [CrossRef]
- Jeltsch, M.; Leppanen, V.M.; Saharinen, P.; Alitalo, K. Receptor tyrosine kinase-mediated angiogenesis. Cold Spring Harb Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Yehya, A.H.S.; Asif, M.; Petersen, S.H.; Subramaniam, A.V.; Kono, K.; Majid, A.M.S.A.; Oon, C.E. Angiogenesis: Managing the culprits behind tumorigenesis and metastasis. Medicina (Kaunas) 2018, 54, 8. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, X.; Kong, B.; Jiang, J. Antiangiogenesis therapy in ovarian cancer patients: An updated meta-analysis for 15 randomized controlled trials. Medicine (Baltimore) 2018, 97, e11920. [Google Scholar] [CrossRef]
- Dal Corso, A.; Pignataro, L.; Belvisi, L.; Gennari, C. αVβ3 Integrin-targeted peptide/peptidomimetic-drug conjugates: In-depth analysis of the linker technology. Curr. Top. Med. Chem. 2016, 16, 314–329. [Google Scholar] [CrossRef]
- Arosio, D.; Manzoni, L.; Corno, C.; Perego, P. Integrin-targeted peptide- and peptidomimetic-drug conjugates for the treatment of tumors. Recent Pat. Anticancer Drug Discov. 2017, 12, 148–168. [Google Scholar] [CrossRef] [PubMed]
- Colombo, R.; Mingozzi, M.; Belvisi, L.; Arosio, D.; Piarulli, U.; Carenini, N.; Perego, P.; Zaffaroni, N.; De Cesare, M.; Castiglioni, V.; et al. Synthesis and biological evaluation (in vitro and in vivo) of cyclic Arginine-Glycine-Aspartate (RGD) peptidomimetic-paclitaxel conjugates targeting integrin αVβ3. J. Med. Chem. 2012, 55, 10460–10474. [Google Scholar] [CrossRef] [PubMed]
- Pilkington-Miksa, M.; Arosio, D.; Belvisi, L.; De Matteo, M.; Vasile, F.; Burreddu, P.; Carta, P.; Rassu, G.; Perego, P.; Carenini, N.; et al. Design, Synthesis and biological evaluation of novel cRGD-Paclitaxel conjugates for integrin-assisted drug delivery. Bioconj. Chem. 2012, 23, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Arosio, D.; Perego, P.; De Cesare, M.; Carenini, N.; Zaffaroni, N.; De Matteo, M.; Manzoni, L. Design, synthesis and biological evaluation of novel dimeric and tetrameric cRGD-Paclitaxel conjugates for integrin-assisted drug delivery. Org. Biomol. Chem. 2015, 13, 7530–7541. [Google Scholar] [CrossRef]
- Zanardi, F.; Burreddu, P.; Rassu, G.; Auzzas, L.; Battistini, L.; Curti, C.; Sartori, A.; Nicastro, G.; Menchi, G.; Cini, N.; et al. Discovery of subnanomolar Arginine-Glycine-Aspartate-based αVβ3/αVβ5 integrin binders embedding 4-aminoproline residues. J. Med. Chem. 2008, 51, 1771–1782. [Google Scholar] [CrossRef]
- Battistini, L.; Burreddu, P.; Carta, P.; Rassu, G.; Auzzas, L.; Curti, C.; Zanardi, F.; Manzoni, L.; Araldi, E.M.V.; Scolastico, C.; et al. 4-Aminoproline-based Arginine-Glycine-Aspartate integrin binders with exposed ligation points: Practical in-solution synthesis, conjugation and binding affinity evaluation. Org. Biomol. Chem. 2009, 7, 4924–4935. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.; Portioli, E.; Battistini, L.; Calorini, L.; Pupi, A.; Vacondio, F.; Arosio, D.; Bianchini, F.; Zanardi, F. Synthesis of novel c(AmpRGD)-sunitinib dual conjugates as molecular tools targeting the αVβ3 integrin/VEGFR2 couple and impairing tumor-associated angiogenesis. J. Med. Chem. 2017, 60, 248–262. [Google Scholar] [CrossRef]
- Bianchini, F.; Portioli, E.; Ferlenghi, F.; Vacondio, F.; Andreucci, E.; Biagioni, A.; Ruzzolini, J.; Peppicelli, S.; Lulli, M.; Calorini, L.; et al. Cell-targeted c(AmpRGD)-sunitinib molecular conjugates impair tumor growth of melanoma. Cancer Lett. 2019, 446, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Somanath, P.R.; Malinin, N.L.; Byzova, T.V. Cooperation between integrin αVβ3 and VEGFR2 in angiogenesis. Angiogenesis 2009, 12, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular pharmacology of VEGF-A isoforms: Binding and signaling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [PubMed]
- Chatzisideri, T.; Leonidis, G.; Sarli, V. Cancer-targeted delivery systems based on peptides. Future Med. Chem. 2018, 10, 2201–2226. [Google Scholar] [CrossRef]
- Srinivasarao, M.; Low, P.S. Ligand-targeted drug delivery. Chem. Rev. 2017, 117, 12133–12164. [Google Scholar] [CrossRef] [PubMed]
- Dolman, M.E.M.; Harmsen, S.; Pieters, E.H.E.; Sparidans, R.W.; Lacombe, M.; Szokol, B.; Orfi, L.; Kéri, G.; Storm, G.; Hennink, W.E.; et al. Targeting a platinum-bound sunitinib analog to renal proximal tubular cells. Int. J. Nanomed. 2012, 7, 417–433. [Google Scholar] [CrossRef]
- Cossa, G.; Gatti, L.; Cassinelli, G.; Lanzi, C.; Zaffaroni, N.; Perego, P. Modulation of sensitivity to antitumor agents by targeting the MAPK survival pathway. Curr. Pharm. Des. 2013, 19, 883–894. [Google Scholar] [CrossRef]
- Sartori, A.; Bianchini, F.; Migliari, S.; Burreddu, P.; Curti, C.; Vacondio, F.; Arosio, D.; Ruffini, L.; Rassu, G.; Calorini, L.; Pupi, A.; Zanardi, F.; Battistini, L. Synthesis and preclinical evaluation of a novel, selective 111In-labelled aminoproline-RGD-peptide for non-invasive melanoma tumor imaging. Med. Chem. Comm. 2015, 6, 2175–2183. [Google Scholar] [CrossRef]
- Corno, C.; Gatti, L.; Arrighetti, N.; Carenini, N.; Zaffaroni, N.; Lanzi, C.; Perego, P. Axl molecular targeting counteracts aggressiveness but not platinum-resistance of ovarian carcinoma cells. Biochem. Pharmacol. 2017, 136, 40–50. [Google Scholar] [CrossRef]
- Imbulgoda, A.; Heng, D.Y.C.; Kollmannsberger, C. Sunitinib in the treatment of advanced solid tumors. In Small Molecules in Oncology. Recent Results in Cancer Research; Martens, U., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 201, pp. 165–184. ISBN 978-3-642-54489-7. [Google Scholar]
- Hao, Z.; Sadek, I. Sunitinib: The antiangiogenic effects and beyond. Onco. Targets Ther. 2016, 9, 5495–5505. [Google Scholar] [CrossRef]
- Cossa, G.; Lanzi, C.; Cassinelli, G.; Carenini, N.; Arrighetti, N.; Gatti, L.; Corna, E.; Zunino, F.; Zaffaroni, N.; Perego, P. Differential outcome of MEK1/2 inhibitor-platinum combinations in platinum-sensitive and -resistant ovarian carcinoma cells. Cancer Lett. 2014, 347, 212–224. [Google Scholar] [CrossRef]
- Perego, P.; Gatti, L.; Righetti, S.C.; Beretta, G.L.; Carenini, N.; Corna, E.; Dal Bo, L.; Tinelli, S.; Colangelo, D.; Leone, R.; et al. Development of resistance to a trinuclear platinum complex in ovarian carcinoma cells. Int. J. Cancer 2003, 105, 617–624. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Days of Treatment | Dose (mg/Kg) | TVI 2 | p3 vs. Controls | BWL% 4 | Tox 5 |
|---|---|---|---|---|---|---|
| Compound 3 | 4–8, 11–15, 18–22, 25–27 | 20 | 60 | 0.0001 | 1 | 0/7 |
| Sunitinib | 4–8, 11–15, 18–22, 25–27 | 40 | 84 | 0.0001 | 0 | 0/7 |
| Cisplatin | 4, 11, 18 | 4.5 | 67 | 0.0001 | 0 | 0/7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sartori, A.; Corno, C.; De Cesare, M.; Scanziani, E.; Minoli, L.; Battistini, L.; Zanardi, F.; Perego, P. Efficacy of a Selective Binder of αVβ3 Integrin Linked to the Tyrosine Kinase Inhibitor Sunitinib in Ovarian Carcinoma Preclinical Models. Cancers 2019, 11, 531. https://doi.org/10.3390/cancers11040531
Sartori A, Corno C, De Cesare M, Scanziani E, Minoli L, Battistini L, Zanardi F, Perego P. Efficacy of a Selective Binder of αVβ3 Integrin Linked to the Tyrosine Kinase Inhibitor Sunitinib in Ovarian Carcinoma Preclinical Models. Cancers. 2019; 11(4):531. https://doi.org/10.3390/cancers11040531
Chicago/Turabian StyleSartori, Andrea, Cristina Corno, Michelandrea De Cesare, Eugenio Scanziani, Lucia Minoli, Lucia Battistini, Franca Zanardi, and Paola Perego. 2019. "Efficacy of a Selective Binder of αVβ3 Integrin Linked to the Tyrosine Kinase Inhibitor Sunitinib in Ovarian Carcinoma Preclinical Models" Cancers 11, no. 4: 531. https://doi.org/10.3390/cancers11040531
APA StyleSartori, A., Corno, C., De Cesare, M., Scanziani, E., Minoli, L., Battistini, L., Zanardi, F., & Perego, P. (2019). Efficacy of a Selective Binder of αVβ3 Integrin Linked to the Tyrosine Kinase Inhibitor Sunitinib in Ovarian Carcinoma Preclinical Models. Cancers, 11(4), 531. https://doi.org/10.3390/cancers11040531