The Role of HOX Transcription Factors in Cancer Predisposition and Progression


Abstract
1. Introduction
2. HOX Transcription Factors in Cancer Predisposition
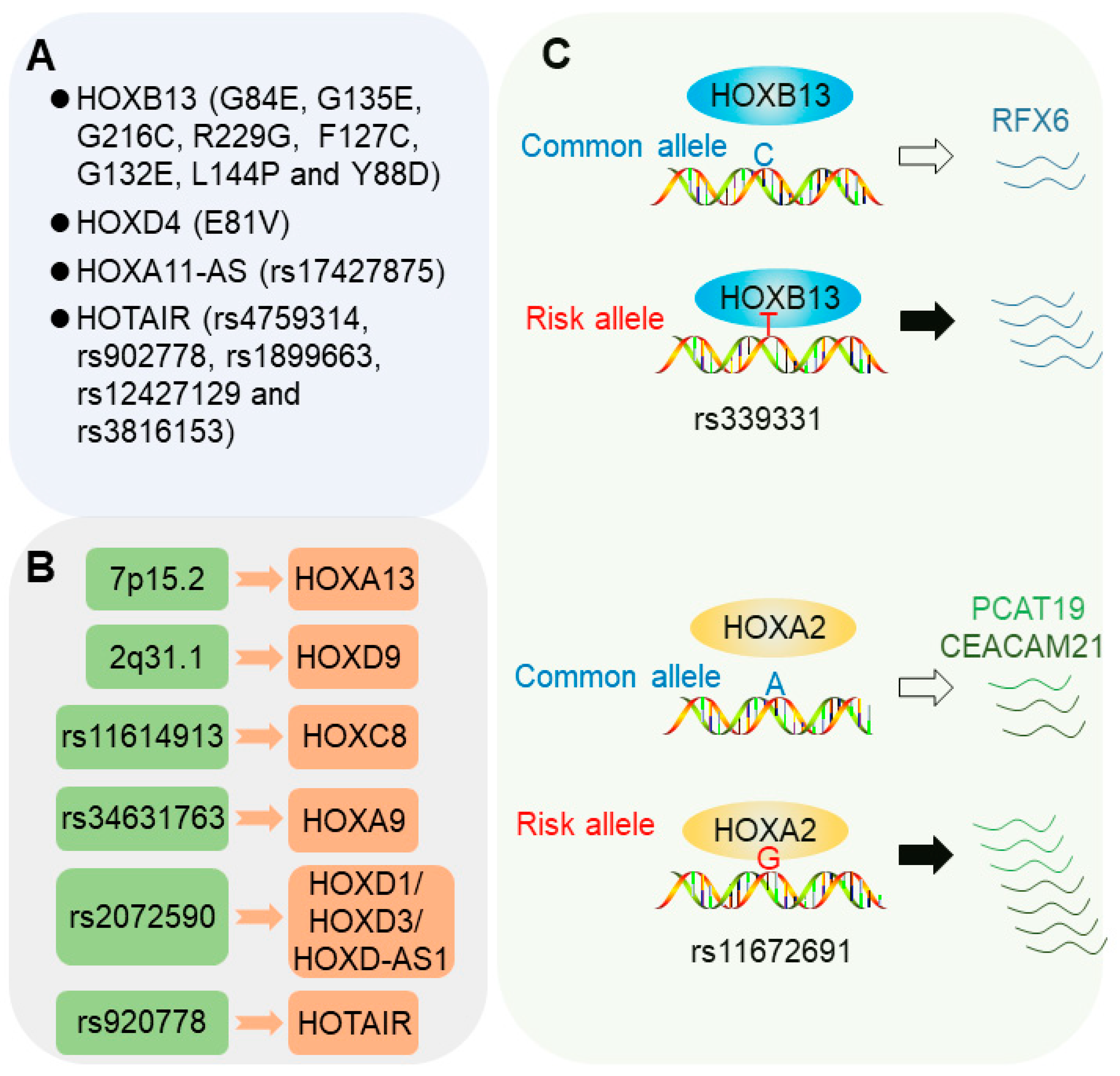
2.1. Coding Genetic Mutations in HOX Genes
2.1.1. HOXB13 Mutations
2.1.2. HOXD4 Mutations
2.1.3. HOX Locus lncRNAs
2.2. Risk Loci Influencing HOX Gene Expression
2.2.1. 7p15.2 Locus
2.2.2. 2q31.1 Locus
2.2.3. 2q31 Allele rs2072590
2.2.4. rs11614913 Locus
2.2.5. rs34631763 Locus
2.2.6. rs920778 Locus
2.3. Risk SNPs Modulating Chromatin Binding of HOX Transcription Factors
2.3.1. 6q22 Allele rs339331
2.3.2. 19q13 Allele rs11672691
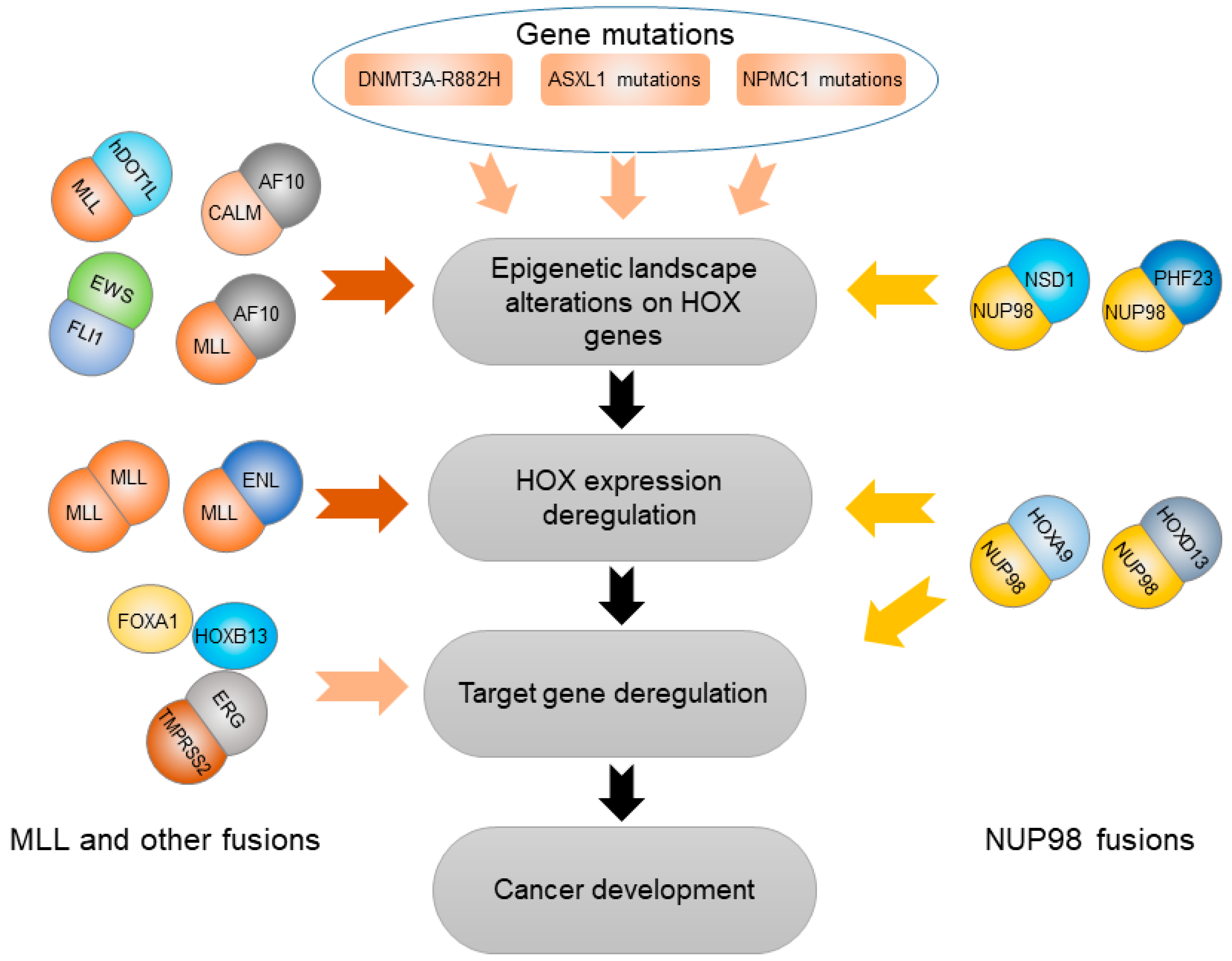
3. HOX Genes Mediate Effects of Other Genetic and Epigenetic Variation
3.1. Abnormal Epigenetic Alteration Affecting HOX Genes
3.2. Somatic Gene Mutations Deregulating HOX Transcription Factors
3.2.1. DNMT3A-R882H
3.2.2. ASXL1 Mutation
3.2.3. NPM1 Mutation
3.3. Gene Fusions Cooperating with HOX Transcription Factors
3.3.1. TMPRSS2-ERG (T2E) Fusion
3.3.2. NUP98 Gene Fusion
3.3.3. MLL and Other Gene Fusions
3.3.4. EWS-FLI1 Fusion
4. HOX Genes in Cancer Progression
4.1. Angiogenesis
4.2. Autophagy
4.3. Differentiation
4.4. Apoptosis
4.5. Proliferation
4.6. Invasion and Metastasis
4.7. Metabolism
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shah, N.; Sukumar, S. The hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010, 10, 361–371. [Google Scholar] [CrossRef]
- Luo, Z.; Rhie, S.K.; Farnham, P.J. The enigmatic hox genes: Can we crack their code? Cancers 2019, 11, 323. [Google Scholar] [CrossRef]
- Berger, M.F.; Badis, G.; Gehrke, A.R.; Talukder, S.; Philippakis, A.A.; Pena-Castillo, L.; Alleyne, T.M.; Mnaimneh, S.; Botvinnik, O.B.; Chan, E.T.; et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 2008, 133, 1266–1276. [Google Scholar] [CrossRef]
- Mann, R.S.; Lelli, K.M.; Joshi, R. Hox specificity unique roles for cofactors and collaborators. Curr. Top. Dev. Biol. 2009, 88, 63–101. [Google Scholar] [PubMed]
- Sun, Y.; Zhou, B.; Mao, F.; Xu, J.; Miao, H.; Zou, Z.; Phuc Khoa, L.T.; Jang, Y.; Cai, S.; Witkin, M.; et al. Hoxa9 reprograms the enhancer landscape to promote leukemogenesis. Cancer Cell 2018, 34, 643–658. [Google Scholar] [CrossRef]
- Mohr, S.; Doebele, C.; Comoglio, F.; Berg, T.; Beck, J.; Bohnenberger, H.; Alexe, G.; Corso, J.; Strobel, P.; Wachter, A.; et al. Hoxa9 and meis1 cooperatively induce addiction to syk signaling by suppressing mir-146a in acute myeloid leukemia. Cancer Cell 2017, 31, 549–562. [Google Scholar] [CrossRef]
- Gilbert, P.M.; Mouw, J.K.; Unger, M.A.; Lakins, J.N.; Gbegnon, M.K.; Clemmer, V.B.; Benezra, M.; Licht, J.D.; Boudreau, N.J.; Tsai, K.K.; et al. Hoxa9 regulates brca1 expression to modulate human breast tumor phenotype. J. Clin. Investig. 2010, 120, 1535–1550. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from sweden, denmark, and finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Peters, U.; Jiao, S.; Schumacher, F.R.; Hutter, C.M.; Aragaki, A.K.; Baron, J.A.; Berndt, S.I.; Bezieau, S.; Brenner, H.; Butterbach, K.; et al. Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology 2013, 144, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Closas, M.; Chanock, S. Genetic susceptibility loci for breast cancer by estrogen receptor status. Clin. Cancer Res. 2008, 14, 8000–8009. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eeles, R.A.; Olama, A.A.; Benlloch, S.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Ghoussaini, M.; Luccarini, C.; Dennis, J.; Jugurnauth-Little, S.; et al. Identification of 23 new prostate cancer susceptibility loci using the icogs custom genotyping array. Nat. Genet. 2013, 45, 385–391. [Google Scholar] [CrossRef]
- Brennan, P.; Hainaut, P.; Boffetta, P. Genetics of lung-cancer susceptibility. Lancet Oncol. 2011, 12, 399–408. [Google Scholar] [CrossRef]
- Chen, D.; Gyllensten, U. Lessons and implications from association studies and post-gwas analyses of cervical cancer. Trends Genet. 2015, 31, 41–54. [Google Scholar] [CrossRef]
- Sherborne, A.L.; Hemminki, K.; Kumar, R.; Bartram, C.R.; Stanulla, M.; Schrappe, M.; Petridou, E.; Semsei, A.F.; Szalai, C.; Sinnett, D.; et al. Rationale for an international consortium to study inherited genetic susceptibility to childhood acute lymphoblastic leukemia. Haematologica 2011, 96, 1049–1054. [Google Scholar] [CrossRef]
- Crowther-Swanepoel, D.; Houlston, R.S. Genetic variation and risk of chronic lymphocytic leukaemia. Semin. Cancer Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef]
- Frank, S.A. Genetic predisposition to cancer—Insights from population genetics. Nat. Rev. Genet. 2004, 5, 764–772. [Google Scholar] [CrossRef]
- Edwards, S.; Campbell, C.; Flohr, P.; Shipley, J.; Giddings, I.; Te-Poele, R.; Dodson, A.; Foster, C.; Clark, J.; Jhavar, S.; et al. Expression analysis onto microarrays of randomly selected cdna clones highlights hoxb13 as a marker of human prostate cancer. Br. J. Cancer 2005, 92, 376–381. [Google Scholar] [CrossRef]
- Wang, Z.; Dahiya, S.; Provencher, H.; Muir, B.; Carney, E.; Coser, K.; Shioda, T.; Ma, X.J.; Sgroi, D.C. The prognostic biomarkers hoxb13, il17br, and chdh are regulated by estrogen in breast cancer. Clin. Cancer Res. 2007, 13, 6327–6334. [Google Scholar] [CrossRef]
- Miao, J.; Wang, Z.; Provencher, H.; Muir, B.; Dahiya, S.; Carney, E.; Leong, C.O.; Sgroi, D.C.; Orsulic, S. Hoxb13 promotes ovarian cancer progression. Proc. Natl. Acad. Sci. USA 2007, 104, 17093–17098. [Google Scholar] [CrossRef]
- Jung, C.; Kim, R.S.; Zhang, H.; Lee, S.J.; Sheng, H.; Loehrer, P.J.; Gardner, T.A.; Jeng, M.H.; Kao, C. Hoxb13 is downregulated in colorectal cancer to confer tcf4-mediated transactivation. Br. J. Cancer 2005, 92, 2233–2239. [Google Scholar] [CrossRef]
- Zhang, E.; Han, L.; Yin, D.; He, X.; Hong, L.; Si, X.; Qiu, M.; Xu, T.; De, W.; Xu, L.; et al. H3k27 acetylation activated-long non-coding rna ccat1 affects cell proliferation and migration by regulating spry4 and hoxb13 expression in esophageal squamous cell carcinoma. Nucleic Acids Res. 2017, 45, 3086–3101. [Google Scholar] [CrossRef]
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline mutations in hoxb13 and prostate-cancer risk. N. Engl. J. Med. 2012, 366, 141–149. [Google Scholar] [CrossRef]
- Lynch, H.T.; Shaw, T.G. Familial prostate cancer and hoxb13 founder mutations: Geographic and racial/ethnic variations. Hum. Genet. 2013, 132, 1–4. [Google Scholar] [CrossRef]
- Karlsson, R.; Aly, M.; Clements, M.; Zheng, L.; Adolfsson, J.; Xu, J.; Gronberg, H.; Wiklund, F. A population-based assessment of germline hoxb13 g84e mutation and prostate cancer risk. Eur. Urol. 2014, 65, 169–176. [Google Scholar] [CrossRef]
- Hoffmann, T.J.; Sakoda, L.C.; Shen, L.; Jorgenson, E.; Habel, L.A.; Liu, J.; Kvale, M.N.; Asgari, M.M.; Banda, Y.; Corley, D.; et al. Imputation of the rare hoxb13 g84e mutation and cancer risk in a large population-based cohort. PLoS Genet. 2015, 11, e1004930. [Google Scholar] [CrossRef]
- Kote-Jarai, Z.; Mikropoulos, C.; Leongamornlert, D.A.; Dadaev, T.; Tymrakiewicz, M.; Saunders, E.J.; Jones, M.; Jugurnauth-Little, S.; Govindasami, K.; Guy, M.; et al. Prevalence of the hoxb13 g84e germline mutation in british men and correlation with prostate cancer risk, tumour characteristics and clinical outcomes. Ann. Oncol. 2015, 26, 756–761. [Google Scholar] [CrossRef]
- Storebjerg, T.M.; Hoyer, S.; Kirkegaard, P.; Bro, F.; LuCamp Study, G.; Orntoft, T.F.; Borre, M.; Sorensen, K.D. Prevalence of the hoxb13 g84e mutation in danish men undergoing radical prostatectomy and its correlations with prostate cancer risk and aggressiveness. BJU Int. 2016, 118, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Johnson, A.M.; Hanson, K.L.; Dayno, M.E.; Kapron, A.L.; Stoffel, E.M.; Cooney, K.A. Germline genetic variants in men with prostate cancer and one or more additional cancers. Cancer 2017, 123, 3925–3932. [Google Scholar] [CrossRef] [PubMed]
- Brechka, H.; Bhanvadia, R.R.; VanOpstall, C.; Vander Griend, D.J. Hoxb13 mutations and binding partners in prostate development and cancer: Function, clinical significance, and future directions. Genes Dis. 2017, 4, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ewing, C.M.; Zheng, S.; Grindedaal, E.M.; Cooney, K.A.; Wiley, K.; Djurovic, S.; Andreassen, O.A.; Axcrona, K.; Mills, I.G.; et al. Genetic factors influencing prostate cancer risk in norwegian men. Prostate 2018, 78, 186–192. [Google Scholar] [CrossRef]
- Lin, X.; Qu, L.; Chen, Z.; Xu, C.; Ye, D.; Shao, Q.; Wang, X.; Qi, J.; Chen, Z.; Zhou, F.; et al. A novel germline mutation in hoxb13 is associated with prostate cancer risk in chinese men. Prostate 2013, 73, 169–175. [Google Scholar] [CrossRef]
- Hayano, T.; Matsui, H.; Nakaoka, H.; Ohtake, N.; Hosomichi, K.; Suzuki, K.; Inoue, I. Germline variants of prostate cancer in japanese families. PLoS ONE 2016, 11, e0164233. [Google Scholar] [CrossRef]
- Beebe-Dimmer, J.L.; Hathcock, M.; Yee, C.; Okoth, L.A.; Ewing, C.M.; Isaacs, W.B.; Cooney, K.A.; Thibodeau, S.N. The hoxb13 g84e mutation is associated with an increased risk for prostate cancer and other malignancies. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1366–1372. [Google Scholar] [CrossRef]
- Alanee, S.; Couch, F.; Offit, K. Association of a hoxb13 variant with breast cancer. N. Engl. J. Med. 2012, 367, 480–481. [Google Scholar] [CrossRef]
- Akbari, M.R.; Anderson, L.N.; Buchanan, D.D.; Clendenning, M.; Jenkins, M.A.; Win, A.K.; Hopper, J.L.; Giles, G.G.; Nam, R.; Narod, S.; et al. Germline hoxb13 p.Gly84glu mutation and risk of colorectal cancer. Cancer Epidemiol. 2013, 37, 424–427. [Google Scholar] [CrossRef]
- Williams, T.M.; Williams, M.E.; Innis, J.W. Range of hox/tale superclass associations and protein domain requirements for hoxa13:Meis interaction. Dev. Biol. 2005, 277, 457–471. [Google Scholar] [CrossRef]
- Johng, D.; Torga, G.; Ewing, C.M.; Jin, K.; Norris, J.D.; McDonnell, D.P.; Isaacs, W.B. Hoxb13 interaction with meis1 modifies proliferation and gene expression in prostate cancer. Prostate 2019, 79, 414–424. [Google Scholar] [CrossRef]
- Chandrasekaran, G.; Hwang, E.C.; Kang, T.W.; Kwon, D.D.; Park, K.; Lee, J.J.; Lakshmanan, V.K. Computational modeling of complete hoxb13 protein for predicting the functional effect of snps and the associated role in hereditary prostate cancer. Sci. Rep. 2017, 7, 43830. [Google Scholar] [CrossRef]
- FitzGerald, L.M.; Raspin, K.; Marthick, J.R.; Field, M.A.; Malley, R.C.; Thomson, R.J.; Blackburn, N.B.; Banks, A.; Charlesworth, J.C.; Donovan, S.; et al. Impact of the G84E variant on HOXB13 gene and protein expression in formalin-fixed, paraffin-embedded prostate tumours. Sci. Rep. 2017, 7, 17778. [Google Scholar] [CrossRef]
- Sipeky, C.; Gao, P.; Zhang, Q.; Wang, L.; Ettala, O.; Talala, K.M.; Tammela, T.L.J.; Auvinen, A.; Wiklund, F.; Wei, G.H.; et al. Synergistic interaction of hoxb13 and cip2a predisposes to aggressive prostate cancer. Clin. Cancer Res. 2018, 24, 6265–6276. [Google Scholar] [CrossRef]
- van Scherpenzeel Thim, V.; Remacle, S.; Picard, J.; Cornu, G.; Gofflot, F.; Rezsohazy, R.; Verellen-Dumoulin, C. Mutation analysis of the hox paralogous 4-13 genes in children with acute lymphoid malignancies: Identification of a novel germline mutation of hoxd4 leading to a partial loss-of-function. Hum. Mutat. 2005, 25, 384–395. [Google Scholar] [CrossRef]
- Richards, E.J.; Permuth-Wey, J.; Li, Y.J.; Chen, Y.A.; Coppola, D.; Reid, B.M.; Lin, H.Y.; Teer, J.K.; Berchuck, A.; Birrer, M.J.; et al. A functional variant in hoxa11-as, a novel long non-coding rna, inhibits the oncogenic phenotype of epithelial ovarian cancer. Oncotarget 2015, 6, 34745–34757. [Google Scholar] [CrossRef]
- Li, J.; Liu, R.; Tang, S.; Feng, F.; Wang, X.; Qi, L.; Liu, C.; Yao, Y.; Sun, C. The effect of long noncoding rnas hox transcript antisense intergenic rna single-nucleotide polymorphisms on breast cancer, cervical cancer, and ovarian cancer susceptibility: A meta-analysis. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, L.; Lin, Z.; Ji, X.; Pi, L.; Lin, X.; Tian, N.; Liu, G.; Liu, Q.; Lin, Z.; et al. Snp-snp and snp-environment interactions of potentially functional hotair snps modify the risk of hepatocellular carcinoma. Mol. Carcinog. 2018. [Google Scholar] [CrossRef]
- Gao, P.; Wei, G.H. Genomic insight into the role of lncrna in cancer susceptibility. Int. J. Mol. Sci. 2017, 18, 1239. [Google Scholar]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.Z.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Huang, Q.; Whitington, T.; Gao, P.; Lindberg, J.F.; Yang, Y.; Sun, J.; Vaisanen, M.R.; Szulkin, R.; Annala, M.; Yan, J.; et al. A prostate cancer susceptibility allele at 6q22 increases rfx6 expression by modulating hoxb13 chromatin binding. Nat. Genet. 2014, 46, 126–135. [Google Scholar] [CrossRef]
- Whitington, T.; Gao, P.; Song, W.; Ross-Adams, H.; Lamb, A.D.; Yang, Y.H.; Svezia, I.; Klevebring, D.; Mills, I.G.; Karlsson, R.; et al. Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nat. Genet. 2016, 48, 387–397. [Google Scholar] [CrossRef]
- Su, J.; Huang, Y.H.; Cui, X.; Wang, X.; Zhang, X.; Lei, Y.; Xu, J.; Lin, X.; Chen, K.; Lv, J.; et al. Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol. 2018, 19, 108. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L.; Fu, Y.P.; Tang, W.; Jacobs, K.B.; Hayes, R.B.; Kraft, P.; Berndt, S.I.; Wacholder, S.; Yu, K.; Hutchinson, A.; et al. Refining the prostate cancer genetic association within the jazf1 gene on chromosome 7p15.2. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1349–1355. [Google Scholar] [CrossRef]
- Chen, L.S.; Fann, J.C.Y.; Chiu, S.Y.H.; Yen, A.M.F.; Wahlfors, T.; Tammela, T.L.; Chen, H.H.; Auvinen, A.; Schleutker, J. Assessing interactions of two loci (rs4242382 and rs10486567) in familial prostate cancer: Statistical evaluation of epistasis. PLoS ONE 2014, 9, e89508. [Google Scholar] [CrossRef]
- Han, Y.; Hazelett, D.J.; Wiklund, F.; Schumacher, F.R.; Stram, D.O.; Berndt, S.I.; Wang, Z.; Rand, K.A.; Hoover, R.N.; Machiela, M.J.; et al. Integration of multiethnic fine-mapping and genomic annotation to prioritize candidate functional snps at prostate cancer susceptibility regions. Hum. Mol. Genet. 2015, 24, 5603–5618. [Google Scholar] [CrossRef]
- Luo, Z.; Rhie, S.K.; Lay, F.D.; Farnham, P.J. A prostate cancer risk element functions as a repressive loop that regulates hoxa13. Cell Rep. 2017, 21, 1411–1417. [Google Scholar] [CrossRef]
- Kelemen, L.E.; Lawrenson, K.; Tyrer, J.; Li, Q.; Lee, J.M.; Seo, J.H.; Phelan, C.M.; Beesley, J.; Chen, X.; Spindler, T.J.; et al. Genome-wide significant risk associations for mucinous ovarian carcinoma. Nat. Genet. 2015, 47, 888–897. [Google Scholar] [CrossRef]
- Lawrenson, K.; Li, Q.; Kar, S.; Seo, J.H.; Tyrer, J.; Spindler, T.J.; Lee, J.; Chen, Y.; Karst, A.; Drapkin, R.; et al. Cis-eqtl analysis and functional validation of candidate susceptibility genes for high-grade serous ovarian cancer. Nat. Commun. 2015, 6, 8234. [Google Scholar] [CrossRef]
- Goode, E.L.; Chenevix-Trench, G.; Song, H.; Ramus, S.J.; Notaridou, M.; Lawrenson, K.; Widschwendter, M.; Vierkant, R.A.; Larson, M.C.; Kjaer, S.K.; et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat. Genet. 2010, 42, 874–879. [Google Scholar] [CrossRef]
- Guo, L.Y.; Peng, Y.; Sun, L.; Han, X.; Xu, J.; Mao, D.W. Ovarian cancer variant rs2072590 is associated with hoxd1 and hoxd3 gene expression. Oncotarget 2017, 8, 103410–103414. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y.; Zhang, X.; Huang, Q.; Diao, Y.; Yin, H.; Liu, H. Long non-coding rna hoxd-as1 in cancer. Clin. Chim. Acta 2018, 487, 197–201. [Google Scholar] [CrossRef]
- Tong, N.; Xu, B.; Shi, D.; Du, M.; Li, X.; Sheng, X.; Wang, M.; Chu, H.; Fang, Y.; Li, J.; et al. Hsa-mir-196a2 polymorphism increases the risk of acute lymphoblastic leukemia in chinese children. Mutat. Res. 2014, 759, 16–21. [Google Scholar] [CrossRef]
- Sibin, M.K.; Harshitha, S.M.; Narasingarao, K.V.; Dhananjaya, I.B.; Dhaval, P.S.; Chetan, G.K. Effect of rs11614913 polymorphism on mature mir196a2 expression and its target gene hoxc8 expression in human glioma. J. Mol. Neurosci. 2017, 61, 144–151. [Google Scholar] [CrossRef]
- Duan, Z.; Zarebski, A.; Montoya-Durango, D.; Grimes, H.L.; Horwitz, M. Gfi1 coordinates epigenetic repression of p21cip/waf1 by recruitment of histone lysine methyltransferase g9a and histone deacetylase 1. Mol. Cell. Biol. 2005, 25, 10338–10351. [Google Scholar] [CrossRef]
- Saleque, S.; Kim, J.; Rooke, H.M.; Orkin, S.H. Epigenetic regulation of hematopoietic differentiation by gfi-1 and gfi-1b is mediated by the cofactors corest and lsd1. Mol. Cell 2007, 27, 562–572. [Google Scholar] [CrossRef]
- Khandanpour, C.; Thiede, C.; Valk, P.J.; Sharif-Askari, E.; Nuckel, H.; Lohmann, D.; Horsthemke, B.; Siffert, W.; Neubauer, A.; Grzeschik, K.H.; et al. A variant allele of growth factor independence 1 (gfi1) is associated with acute myeloid leukemia. Blood 2010, 115, 2462–2472. [Google Scholar] [CrossRef]
- Khandanpour, C.; Krongold, J.; Schutte, J.; Bouwman, F.; Vassen, L.; Gaudreau, M.C.; Chen, R.; Calero-Nieto, F.J.; Diamanti, E.; Hannah, R.; et al. The human gfi136n variant induces epigenetic changes at the hoxa9 locus and accelerates k-ras driven myeloproliferative disorder in mice. Blood 2012, 120, 4006–4017. [Google Scholar] [CrossRef]
- Botezatu, L.; Michel, L.C.; Helness, A.; Vadnais, C.; Makishima, H.; Hones, J.M.; Robert, F.; Vassen, L.; Thivakaran, A.; Al-Matary, Y.; et al. Epigenetic therapy as a novel approach for gfi136n-associated murine/human aml. Exp. Hematol. 2016, 44, 713–726. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, L.; Fu, G.; Sun, F.; Shi, J.; Wei, J.; Lu, C.; Zhou, C.; Yuan, Q.; Yang, M. The identification of an escc susceptibility snp rs920778 that regulates the expression of lncrna hotair via a novel intronic enhancer. Carcinogenesis 2014, 35, 2062–2067. [Google Scholar] [CrossRef]
- Deplancke, B.; Alpern, D.; Gardeux, V. The genetics of transcription factor DNA binding variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef]
- Gao, P.; Xia, J.H.; Sipeky, C.; Dong, X.M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and clinical implications of the 19q13 aggressive prostate cancer susceptibility locus. Cell 2018, 174, 576–589. [Google Scholar] [CrossRef]
- Spisak, S.; Lawrenson, K.; Fu, Y.; Csabai, I.; Cottman, R.T.; Seo, J.H.; Haiman, C.; Han, Y.; Lenci, R.; Li, Q.; et al. Causel: An epigenome- and genome-editing pipeline for establishing function of noncoding gwas variants. Nat. Med. 2015, 21, 1357–1363. [Google Scholar] [CrossRef]
- Amin Al Olama, A.; Kote-Jarai, Z.; Schumacher, F.R.; Wiklund, F.; Berndt, S.I.; Benlloch, S.; Giles, G.G.; Severi, G.; Neal, D.E.; Hamdy, F.C.; et al. A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum. Mol. Genet. 2013, 22, 408–415. [Google Scholar] [CrossRef]
- Shui, I.M.; Lindstrom, S.; Kibel, A.S.; Berndt, S.I.; Campa, D.; Gerke, T.; Penney, K.L.; Albanes, D.; Berg, C.; Bueno-de-Mesquita, H.B.; et al. Prostate cancer (pca) risk variants and risk of fatal pca in the national cancer institute breast and prostate cancer cohort consortium. Eur. Urol. 2014, 65, 1069–1075. [Google Scholar] [CrossRef]
- Hua, J.T.; Ahmed, M.; Guo, H.; Zhang, Y.; Chen, S.; Soares, F.; Lu, J.; Zhou, S.; Wang, M.; Li, H.; et al. Risk snp-mediated promoter-enhancer switching drives prostate cancer through lncrna pcat19. Cell 2018, 174, 564–575. [Google Scholar] [CrossRef]
- Xia, J.H.; Wei, G.H. Oncogenic regulatory circuits driven by 19q13 rs11672691 underlies prostate cancer aggressiveness. Mol. Cell. Oncol. 2018, 5, e1516451. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Strathdee, G.; Brown, R. Aberrant DNA methylation in cancer: Potential clinical interventions. Expert Rev. Mol. Med. 2002, 4, 1–17. [Google Scholar] [CrossRef]
- Chen, L.N.; Rubin, R.S.; Othepa, E.; Cer, C.; Yun, E.; Agarwal, R.P.; Collins, B.T.; McGeagh, K.; Pahira, J.; Bandi, G.; et al. Correlation of hoxd3 promoter hypermethylation with clinical and pathologic features in screening prostate biopsies. Prostate 2014, 74, 714–721. [Google Scholar] [CrossRef][Green Version]
- Pilato, B.; Pinto, R.; De Summa, S.; Lambo, R.; Paradiso, A.; Tommasi, S. Hox gene methylation status analysis in patients with hereditary breast cancer. J. Hum. Genet. 2013, 58, 51–53. [Google Scholar] [CrossRef][Green Version]
- Pfeifer, G.P.; Rauch, T.A. DNA methylation patterns in lung carcinomas. Semin. Cancer Biol. 2009, 19, 181–187. [Google Scholar] [CrossRef]
- Karlsson, A.; Jonsson, M.; Lauss, M.; Brunnstrom, H.; Jonsson, P.; Borg, A.; Jonsson, G.; Ringner, M.; Planck, M.; Staaf, J. Genome-wide DNA methylation analysis of lung carcinoma reveals one neuroendocrine and four adenocarcinoma epitypes associated with patient outcome. Clin. Cancer Res. 2014, 20, 6127–6140. [Google Scholar] [CrossRef]
- Strathdee, G.; Sim, A.; Parker, A.; Oscier, D.; Brown, R. Promoter hypermethylation silences expression of the hoxa4 gene and correlates with igvh mutational status in cll. Leukemia 2006, 20, 1326–1329. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Lee, S.H.; Jones, A.; Fiegl, H.; Kalwa, M.; Wagner, W.; Chindera, K.; Evans, I.; Dubeau, L.; Orjalo, A.; et al. Hotair and its surrogate DNA methylation signature indicate carboplatin resistance in ovarian cancer. Genome Med. 2015, 7, 108. [Google Scholar] [CrossRef]
- Liu, Y.W.; Sun, M.; Xia, R.; Zhang, E.B.; Liu, X.H.; Zhang, Z.H.; Xu, T.P.; De, W.; Liu, B.R.; Wang, Z.X. Linchotair epigenetically silences mir34a by binding to prc2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015, 6, e1802. [Google Scholar] [CrossRef]
- Xylinas, E.; Hassler, M.R.; Zhuang, D.; Krzywinski, M.; Erdem, Z.; Robinson, B.D.; Elemento, O.; Clozel, T.; Shariat, S.F. An epigenomic approach to improving response to neoadjuvant cisplatin chemotherapy in bladder cancer. Biomolecules 2016, 6, 37. [Google Scholar] [CrossRef]
- Mayle, A.; Yang, L.; Rodriguez, B.; Zhou, T.; Chang, E.; Curry, C.V.; Challen, G.A.; Li, W.; Wheeler, D.; Rebel, V.I.; et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015, 125, 629–638. [Google Scholar] [CrossRef]
- Tan, Y.T.; Sun, Y.; Zhu, S.H.; Ye, L.; Zhao, C.J.; Zhao, W.L.; Chen, Z.; Chen, S.J.; Liu, H. Deregulation of hox genes by dnmt3a and mll mutations converges on bmi1. Leukemia 2016, 30, 1609–1612. [Google Scholar] [CrossRef]
- Lu, R.; Wang, P.; Parton, T.; Zhou, Y.; Chrysovergis, K.; Rockowitz, S.; Chen, W.Y.; Abdel-Wahab, O.; Wade, P.A.; Zheng, D.; et al. Epigenetic perturbations by arg882-mutated dnmt3a potentiate aberrant stem cell gene-expression program and acute leukemia development. Cancer Cell 2016, 30, 92–107. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. Asxl1 mutations promote myeloid transformation through loss of prc2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Vassiliou, G.S.; Cooper, J.L.; Rad, R.; Li, J.; Rice, S.; Uren, A.; Rad, L.; Ellis, P.; Andrews, R.; Banerjee, R.; et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat. Genet. 2011, 43, 470–475. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant npm1 maintains the leukemic state through hox expression. Cancer Cell 2018, 34, 499–512. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of tmprss2 and ets transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.J.; Fraser, M.; van der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. Tmprss2-erg fusion co-opts master transcription factors and activates notch signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef]
- Wang, J.; Cai, Y.; Ren, C.; Ittmann, M. Expression of variant tmprss2/erg fusion messenger rnas is associated with aggressive prostate cancer. Cancer Res. 2006, 66, 8347–8351. [Google Scholar] [CrossRef]
- Gough, S.M.; Lee, F.; Yang, F.; Walker, R.L.; Zhu, Y.J.; Pineda, M.; Onozawa, M.; Chung, Y.J.; Bilke, S.; Wagner, E.K.; et al. Nup98-phf23 is a chromatin-modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of phd histone reader function. Cancer Discov. 2014, 4, 564–577. [Google Scholar] [CrossRef]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. Nup98 gene fusions and hematopoietic malignancies: Common themes and new biologic insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef]
- Xu, H.; Valerio, D.G.; Eisold, M.E.; Sinha, A.; Koche, R.P.; Hu, W.; Chen, C.W.; Chu, S.H.; Brien, G.L.; Park, C.Y.; et al. Nup98 fusion proteins interact with the nsl and mll1 complexes to drive leukemogenesis. Cancer Cell 2016, 30, 863–878. [Google Scholar] [CrossRef]
- Takeda, A.; Goolsby, C.; Yaseen, N.R. Nup98-hoxa9 induces long-term proliferation and blocks differentiation of primary human cd34+ hematopoietic cells. Cancer Res. 2006, 66, 6628–6637. [Google Scholar] [CrossRef]
- Shields, B.J.; Slape, C.I.; Vo, N.; Jackson, J.T.; Pliego-Zamora, A.; Ranasinghe, H.; Shi, W.; Curtis, D.J.; McCormack, M.P. The nup98-hoxd13 fusion oncogene induces thymocyte self-renewal via lmo2/lyl1. Leukemia 2019. [Google Scholar] [CrossRef]
- McCormack, M.P.; Shields, B.J.; Jackson, J.T.; Nasa, C.; Shi, W.; Slater, N.J.; Tremblay, C.S.; Rabbitts, T.H.; Curtis, D.J. Requirement for Lyl1 in a model of Lmo2-driven early T-cell precursor ALL. Blood 2013, 122, 2093–2103. [Google Scholar] [CrossRef]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. Nup98-nsd1 links h3k36 methylation to hox-a gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef]
- Muntean, A.G.; Hess, J.L. The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol. 2012, 7, 283–301. [Google Scholar] [CrossRef]
- Ayton, P.M.; Cleary, M.L. Transformation of myeloid progenitors by mll oncoproteins is dependent on hoxa7 and hoxa9. Genes Dev. 2003, 17, 2298–2307. [Google Scholar] [CrossRef]
- Horton, S.J.; Grier, D.G.; McGonigle, G.J.; Thompson, A.; Morrow, M.; De Silva, I.; Moulding, D.A.; Kioussis, D.; Lappin, T.R.; Brady, H.J.; et al. Continuous mll-enl expression is necessary to establish a “hox code” and maintain immortalization of hematopoietic progenitor cells. Cancer Res. 2005, 65, 9245–9252. [Google Scholar] [CrossRef]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. Hdot1l links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, B.; Qin, S.; Xu, J.; Harding, R.; Tempel, W.; Nayak, V.; Li, Y.; Loppnau, P.; Dou, Y.; et al. Structural and functional analysis of the dot1l-af10 complex reveals mechanistic insights into mll-af10-associated leukemogenesis. Genes Dev. 2018, 32, 341–346. [Google Scholar] [CrossRef]
- Lin, Y.H.; Kakadia, P.M.; Chen, Y.; Li, Y.Q.; Deshpande, A.J.; Buske, C.; Zhang, K.L.; Zhang, Y.; Xu, G.L.; Bohlander, S.K. Global reduction of the epigenetic h3k79 methylation mark and increased chromosomal instability in calm-af10-positive leukemias. Blood 2009, 114, 651–658. [Google Scholar] [CrossRef]
- Hsu, K.; Look, A.T. Turning on a dimer. Cancer Cell 2003, 4, 81–83. [Google Scholar] [CrossRef]
- Pfaltzgraff, E.R.; Apfelbaum, A.; Kassa, A.P.; Song, J.Y.; Jiang, W.; Suhan, T.K.; Wellik, D.M.; Lawlor, E.R. Anatomic origin of osteochondrogenic progenitors impacts sensitivity to ews-fli1-induced transformation. Cancers 2019, 11, 313. [Google Scholar] [CrossRef]
- Svoboda, L.K.; Harris, A.; Bailey, N.J.; Schwentner, R.; Tomazou, E.; von Levetzow, C.; Magnuson, B.; Ljungman, M.; Kovar, H.; Lawlor, E.R. Overexpression of hox genes is prevalent in ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics 2014, 9, 1613–1625. [Google Scholar] [CrossRef]
- Care, A.; Felicetti, F.; Meccia, E.; Bottero, L.; Parenza, M.; Stoppacciaro, A.; Peschle, C.; Colombo, M.P. Hoxb7: A key factor for tumor-associated angiogenic switch. Cancer Res. 2001, 61, 6532–6539. [Google Scholar]
- Storti, P.; Donofrio, G.; Colla, S.; Airoldi, I.; Bolzoni, M.; Agnelli, L.; Abeltino, M.; Todoerti, K.; Lazzaretti, M.; Mancini, C.; et al. Hoxb7 expression by myeloma cells regulates their pro-angiogenic properties in multiple myeloma patients. Leukemia 2011, 25, 527–537. [Google Scholar] [CrossRef]
- Heinonen, H.; Lepikhova, T.; Sahu, B.; Pehkonen, H.; Pihlajamaa, P.; Louhimo, R.; Gao, P.; Wei, G.H.; Hautaniemi, S.; Janne, O.A.; et al. Identification of several potential chromatin binding sites of hoxb7 and its downstream target genes in breast cancer. Int. J. Cancer 2015, 137, 2374–2383. [Google Scholar] [CrossRef]
- Wu, S.Y.; Rupaimoole, R.; Shen, F.; Pradeep, S.; Pecot, C.V.; Ivan, C.; Nagaraja, A.S.; Gharpure, K.M.; Pham, E.; Hatakeyama, H.; et al. A mir-192-egr1-hoxb9 regulatory network controls the angiogenic switch in cancer. Nat. Commun. 2016, 7, 11169. [Google Scholar] [CrossRef]
- Hayashida, T.; Takahashi, F.; Chiba, N.; Brachtel, E.; Takahashi, M.; Godin-Heymann, N.; Gross, K.W.; Vivanco, M.; Wijendran, V.; Shioda, T.; et al. Hoxb9, a gene overexpressed in breast cancer, promotes tumorigenicity and lung metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1100–1105. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, W.J.; Gan, T.Q.; Zhang, X.L.; Xie, Z.C.; Ye, Z.H.; Deng, Y.; Wang, Z.F.; Cai, K.T.; Li, S.K.; et al. Clinical significance and effect of lncrna hoxa11-as in nsclc: A study based on bioinformatics, in vitro and in vivo verification. Sci. Rep. 2017, 7, 5567. [Google Scholar] [CrossRef]
- Chen, Y.; Gorski, D.H. Regulation of angiogenesis through a microrna (mir-130a) that down-regulates antiangiogenic homeobox genes gax and hoxa5. Blood 2007, 111, 1217–1226. [Google Scholar] [CrossRef]
- Rhoads, K.; Arderiu, G.; Charboneau, A.; Hansen, S.L.; Hoffman, W.; Boudreau, N. A role for hox a5 in regulating angiogenesis and vascular patterning. Lymphat. Res. Biol. 2005, 3, 240–252. [Google Scholar] [CrossRef]
- Xuan, F.; Huang, M.; Liu, W.; Ding, H.; Yang, L.; Cui, H. Homeobox c9 suppresses beclin1-mediated autophagy in glioblastoma by directly inhibiting the transcription of death-associated protein kinase 1. Neuro Oncol. 2016, 18, 819–829. [Google Scholar] [CrossRef]
- Tsai, K.W.; Leung, C.M.; Lo, Y.H.; Chen, T.W.; Chan, W.C.; Yu, S.Y.; Tu, Y.T.; Lam, H.C.; Li, S.C.; Ger, L.P.; et al. Arm selection preference of microrna-193a varies in breast cancer. Sci. Rep. 2016, 6, 28176. [Google Scholar] [CrossRef]
- Cheng, J.Z.; Chen, J.J.; Wang, Z.G.; Yu, D. Microrna-185 inhibits cell proliferation while promoting apoptosis and autophagy through negative regulation of tgf-beta 1/mtor axis and hoxc6 in nasopharyngeal carcinoma. Cancer Biomark. 2018, 23, 107–123. [Google Scholar] [CrossRef]
- Pawlowska, E.; Szczepanska, J.; Blasiak, J. The long noncoding rna hotair in breast cancer: Does autophagy play a role? Int. J. Mol. Sci. 2017, 18, 2317. [Google Scholar] [CrossRef]
- Guo, X.; Xiao, H.; Guo, S.; Li, J.; Wang, Y.; Chen, J.; Lou, G. Long noncoding rna hotair knockdown inhibits autophagy and epithelial-mesenchymal transition through the wnt signaling pathway in radioresistant human cervical cancer hela cells. J. Cell. Physiol. 2019, 234, 3478–3489. [Google Scholar] [CrossRef]
- Bach, C.; Buhl, S.; Mueller, D.; Garcia-Cuellar, M.P.; Maethner, E.; Slany, R.K. Leukemogenic transformation by hoxa cluster genes. Blood 2010, 115, 2910–2918. [Google Scholar] [CrossRef]
- Lai, C.K.; Norddahl, G.L.; Maetzig, T.; Rosten, P.; Lohr, T.; Sanchez Milde, L.; von Krosigk, N.; Docking, T.R.; Heuser, M.; Karsan, A.; et al. Meis2 as a critical player in mn1-induced leukemia. Blood Cancer J. 2017, 7, e613. [Google Scholar] [CrossRef]
- Wermuth, P.J.; Buchberg, A.M. Meis1-mediated apoptosis is caspase dependent and can be suppressed by coexpression of hoxa9 in murine and human cell lines. Blood 2005, 105, 1222–1230. [Google Scholar] [CrossRef]
- Somerville, T.D.; Wiseman, D.H.; Spencer, G.J.; Huang, X.; Lynch, J.T.; Leong, H.S.; Williams, E.L.; Cheesman, E.; Somervaille, T.C. Frequent derepression of the mesenchymal transcription factor gene foxc1 in acute myeloid leukemia. Cancer Cell 2015, 28, 329–342. [Google Scholar] [CrossRef]
- Hatanaka, Y.; de Velasco, M.A.; Oki, T.; Shimizu, N.; Nozawa, M.; Yoshimura, K.; Yoshikawa, K.; Nishio, K.; Uemura, H. Hoxa10 expression profiling in prostate cancer. Prostate 2019, 79, 554–563. [Google Scholar] [CrossRef]
- Tanwar, P.S.; Kaneko-Tarui, T.; Lee, H.J.; Zhang, L.H.; Teixeira, J.M. Pten loss and hoxa10 expression are associated with ovarian endometrioid adenocarcinoma differentiation and progression. Carcinogenesis 2013, 34, 893–901. [Google Scholar] [CrossRef]
- Krishnaraju, K.; Hoffman, B.; Liebermann, D.A. Lineage-specific regulation of hematopoiesis by hox-b8 (hox-2.4): Inhibition of granulocytic differentiation and potentiation of monocytic differentiation. Blood 1997, 90, 1840–1849. [Google Scholar]
- Mao, L.; Ding, J.; Zha, Y.; Yang, L.; McCarthy, B.A.; King, W.; Cui, H.; Ding, H.F. Hoxc9 links cell-cycle exit and neuronal differentiation and is a prognostic marker in neuroblastoma. Cancer Res. 2011, 71, 4314–4324. [Google Scholar] [CrossRef]
- Tan, S.H.; Barker, N. Stemming colorectal cancer growth and metastasis: Hoxa5 forces cancer stem cells to differentiate. Cancer Cell 2015, 28, 683–685. [Google Scholar] [CrossRef]
- Ordonez-Moran, P.; Dafflon, C.; Imajo, M.; Nishida, E.; Huelsken, J. Hoxa5 counteracts stem cell traits by inhibiting wnt signaling in colorectal cancer. Cancer Cell 2015, 28, 815–829. [Google Scholar] [CrossRef]
- Heubach, J.; Monsior, J.; Deenen, R.; Niegisch, G.; Szarvas, T.; Niedworok, C.; Schulz, W.A.; Hoffmann, M.J. The long noncoding rna hotair has tissue and cell type-dependent effects on hox gene expression and phenotype of urothelial cancer cells. Mol. Cancer 2015, 14, 108. [Google Scholar] [CrossRef]
- Raman, V.; Martensen, S.A.; Reisman, D.; Evron, E.; Odenwald, W.F.; Jaffee, E.; Marks, J.; Sukumar, S. Compromised hoxa5 function can limit p53 expression in human breast tumours. Nature 2000, 405, 974–978. [Google Scholar] [CrossRef]
- Chen, H.; Chung, S.; Sukumar, S. Hoxa5-induced apoptosis in breast cancer cells is mediated by caspases 2 and 8. Mol. Cell. Biol. 2003, 24, 924–935. [Google Scholar] [CrossRef]
- Liu, W.J.; Zhang, T.; Guo, Q.L.; Liu, C.Y.; Bai, Y.Q. Effect of atra on the expression of hoxa5 gene in k562 cells and its relationship with cell cycle and apoptosis. Mol. Med. Rep. 2016, 13, 4221–4228. [Google Scholar] [CrossRef]
- Huang, H.P.; Liu, W.J.; Guo, Q.L.; Bai, Y.Q. Effect of silencing hoxa5 gene expression using rna interference on cell cycle and apoptosis in jurkat cells. Int. J. Mol. Med. 2016, 37, 669–678. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Yang, T.Q.; Zhou, B.; Yang, M.X.; Feng, H.J.; Wang, Y.L. Hoxa5 overexpression promotes osteosarcoma cell apoptosis through the p53 and p38alpha mapk pathway. Gene 2018, 689, 18–23. [Google Scholar] [CrossRef]
- Zhu, Q.; Lv, T.; Wu, Y.; Shi, X.; Liu, H.; Song, Y. Long non-coding rna 00312 regulated by hoxa5 inhibits tumour proliferation and promotes apoptosis in non-small cell lung cancer. J. Cell. Mol. Med. 2017, 21, 2184–2198. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, C.; Wang, H. Hoxa5 inhibits the proliferation and induces the apoptosis of cervical cancer cells via regulation of protein kinase b and p27. Oncol Rep. 2019, 41, 1122–1130. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, H.; Lee, J.; Liang, X.; Wu, X.; Zhu, T.; Lo, P.K.; Zhang, X.; Sukumar, S. Hoxa5 acts directly downstream of retinoic acid receptor beta and contributes to retinoic acid-induced apoptosis and growth inhibition. Cancer Res. 2007, 67, 8007–8013. [Google Scholar] [CrossRef]
- Chu, M.C.; Selam, F.B.; Taylor, H.S. Hoxa10 regulates p53 expression and matrigel invasion in human breast cancer cells. Cancer Biol. Ther. 2014, 3, 568–572. [Google Scholar] [CrossRef]
- Moon, S.M.; Kim, S.A.; Yoon, J.H.; Ahn, S.G. Hoxc6 is deregulated in human head and neck squamous cell carcinoma and modulates bcl-2 expression. J. Biol. Chem. 2012, 287, 35678–35688. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Liu, N.; Hou, J.; Xiao, W.; Wang, H. Hoxc6 promotes cervical cancer progression via regulation of bcl-2. FASEB J. 2019, 33, 3901–3911. [Google Scholar] [CrossRef]
- Ramachandran, S.; Liu, P.; Young, A.N.; Yin-Goen, Q.; Lim, S.D.; Laycock, N.; Amin, M.B.; Carney, J.K.; Marshall, F.F.; Petros, J.A.; et al. Loss of hoxc6 expression induces apoptosis in prostate cancer cells. Oncogene 2005, 24, 188–198. [Google Scholar] [CrossRef]
- de Bock, C.E.; Demeyer, S.; Degryse, S.; Verbeke, D.; Sweron, B.; Gielen, O.; Vandepoel, R.; Vicente, C.; Vanden Bempt, M.; Dagklis, A.; et al. Hoxa9 cooperates with activated jak/stat signaling to drive leukemia development. Cancer Discov. 2018, 8, 616–631. [Google Scholar] [CrossRef]
- Brock, A.; Krause, S.; Li, H.; Kowalski, M.; Goldberg, M.S.; Collins, J.J.; Ingber, D.E. Silencing hoxa1 by intraductal injection of sirna lipidoid nanoparticles prevents mammary tumor progression in mice. Sci. Transl. Med. 2014, 6, 217ra2. [Google Scholar] [CrossRef]
- Taminiau, A.; Draime, A.; Tys, J.; Lambert, B.; Vandeputte, J.; Nguyen, N.; Renard, P.; Geerts, D.; Rezsohazy, R. Hoxa1 binds rbck1/hoil-1 and traf2 and modulates the tnf/nf-kappab pathway in a transcription-independent manner. Nucleic Acids Res. 2016, 44, 7331–7349. [Google Scholar]
- Steger, J.; Fuller, E.; Garcia-Cuellar, M.P.; Hetzner, K.; Slany, R.K. Insulin-like growth factor 1 is a direct hoxa9 target important for hematopoietic transformation. Leukemia 2015, 29, 901–908. [Google Scholar] [CrossRef]
- Liao, W.T.; Jiang, D.; Yuan, J.; Cui, Y.M.; Shi, X.W.; Chen, C.M.; Bian, X.W.; Deng, Y.J.; Ding, Y.Q. Hoxb7 as a prognostic factor and mediator of colorectal cancer progression. Clin. Cancer Res. 2011, 17, 3569–3578. [Google Scholar] [CrossRef]
- Huan, H.B.; Yang, D.P.; Wen, X.D.; Chen, X.J.; Zhang, L.; Wu, L.L.; Bie, P.; Xia, F. Hoxb7 accelerates the malignant progression of hepatocellular carcinoma by promoting stemness and epithelial-mesenchymal transition. J. Exp. Clin. Cancer Res. 2017, 36, 86. [Google Scholar] [CrossRef]
- Fujiki, K.; Duerr, E.M.; Kikuchi, H.; Ng, A.; Xavier, R.J.; Mizukami, Y.; Imamura, T.; Kulke, M.H.; Chung, D.C. Hoxc6 is overexpressed in gastrointestinal carcinoids and interacts with jund to regulate tumor growth. Gastroenterology 2008, 135, 907–916. [Google Scholar] [CrossRef]
- Palakurthy, R.K.; Wajapeyee, N.; Santra, M.K.; Gazin, C.; Lin, L.; Gobeil, S.; Green, M.R. Epigenetic silencing of the rassf1a tumor suppressor gene through hoxb3-mediated induction of dnmt3b expression. Mol. Cell 2009, 36, 219–230. [Google Scholar] [CrossRef]
- Wang, L.M.; Sun, H.F.; Wang, X.F.; Hou, N.; Zhao, L.Y.; Tong, D.D.; He, K.; Yang, Y.; Song, T.S.; Yang, J.; et al. Egr1 mediates mir-203a suppress the hepatocellular carcinoma cells progression by targeting hoxd3 through egfr signaling pathway. Oncotarget 2016, 7, 45302–45316. [Google Scholar] [CrossRef]
- Nagel, S.; Burek, C.; Venturini, L.; Scherr, M.; Quentmeier, H.; Meyer, C.; Rosenwald, A.; Drexler, H.G.; MacLeod, R.A. Comprehensive analysis of homeobox genes in hodgkin lymphoma cell lines identifies dysregulated expression of hoxb9 mediated via erk5 signaling and bmi1. Blood 2007, 109, 3015–3023. [Google Scholar] [CrossRef]
- Yan, T.; Ooi, W.F.; Qamra, A.; Cheung, A.; Ma, D.; Sundaram, G.M.; Xu, C.; Xing, M.; Poon, L.; Wang, J.; et al. Hoxc5 and mir-615-3p target newly evolved genomic regions to repress htert and inhibit tumorigenesis. Nat. Commun. 2018, 9, 100. [Google Scholar] [CrossRef]
- Hahn, W.C.; Stewart, S.A.; Brooks, M.W.; York, S.G.; Eaton, E.; Kurachi, A.; Beijersbergen, R.L.; Knoll, J.H.M.; Meyerson, M.; Weinberg, R.A. Inhibition of telomerase limits the growth of human cancer cells. Nat. Med. 1999, 5, 1164–1170. [Google Scholar] [CrossRef]
- Bjornsson, J.M.; Andersson, E.; Lundstrom, P.; Larsson, N.; Xu, X.F.; Repetowska, E.; Humphries, R.K.; Karlsson, S. Proliferation of primitive myeloid progenitors can be reversibly induced by hoxa10. Blood 2001, 98, 3301–3308. [Google Scholar] [CrossRef]
- Chen, R.; Li, H.; Li, Y.; Fazli, L.; Gleave, M.; Nappi, L.; Dong, X. Loss of nuclear functions of hoxa10 is associated with testicular cancer proliferation. Front. Oncol. 2018, 8, 594. [Google Scholar] [CrossRef]
- Sun, M.; Song, C.X.; Huang, H.; Frankenberger, C.A.; Sankarasharma, D.; Gomes, S.; Chen, P.; Chen, J.; Chada, K.K.; He, C.; et al. Hmga2/tet1/hoxa9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 9920–9925. [Google Scholar] [CrossRef]
- Yoshida, H.; Broaddus, R.; Cheng, W.; Xie, S.; Naora, H. Deregulation of the hoxa10 homeobox gene in endometrial carcinoma: Role in epithelial-mesenchymal transition. Cancer Res. 2006, 66, 889–897. [Google Scholar] [CrossRef]
- Weiss, F.U.; Marques, I.J.; Woltering, J.M.; Vlecken, D.H.; Aghdassi, A.; Partecke, L.I.; Heidecke, C.D.; Lerch, M.M.; Bagowski, C.P. Retinoic acid receptor antagonists inhibit mir-10a expression and block metastatic behavior of pancreatic cancer. Gastroenterology 2009, 137, 2136–2145. [Google Scholar] [CrossRef]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of mir-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef]
- Kim, J.; Siverly, A.N.; Chen, D.; Wang, M.; Yuan, Y.; Wang, Y.; Lee, H.; Zhang, J.; Muller, W.J.; Liang, H.; et al. Ablation of mir-10b suppresses oncogene-induced mammary tumorigenesis and metastasis and reactivates tumor-suppressive pathways. Cancer Res. 2016, 76, 6424–6435. [Google Scholar] [CrossRef] [PubMed]
- Gabriely, G.; Teplyuk, N.M.; Krichevsky, A.M. Context effect: Microrna-10b in cancer cell proliferation, spread and death. Autophagy 2011, 7, 1384–1386. [Google Scholar] [CrossRef]
- Reddy, S.D.; Ohshiro, K.; Rayala, S.K.; Kumar, R. Microrna-7, a homeobox d10 target, inhibits p21-activated kinase 1 and regulates its functions. Cancer Res. 2008, 68, 8195–8200. [Google Scholar] [CrossRef]
- Wu, X.; Chen, H.; Parker, B.; Rubin, E.; Zhu, T.; Lee, J.S.; Argani, P.; Sukumar, S. Hoxb7, a homeodomain protein, is overexpressed in breast cancer and confers epithelial-mesenchymal transition. Cancer Res. 2006, 66, 9527–9534. [Google Scholar] [CrossRef]
- Liu, S.; Jin, K.; Hui, Y.; Fu, J.; Jie, C.; Feng, S.; Reisman, D.; Wang, Q.; Fan, D.; Sukumar, S.; et al. Hoxb7 promotes malignant progression by activating the tgfbeta signaling pathway. Cancer Res. 2015, 75, 709–719. [Google Scholar] [CrossRef]
- Stiegelbauer, V.; Vychytilova-Faltejskova, P.; Karbiener, M.; Pehserl, A.M.; Reicher, A.; Resel, M.; Heitzer, E.; Ivan, C.; Bullock, M.; Ling, H.; et al. Mir-196b-5p regulates colorectal cancer cell migration and metastases through interaction with hoxb7 and galnt5. Clin. Cancer Res. 2017, 23, 5255–5266. [Google Scholar] [CrossRef]
- Sun, M.; Nie, F.; Wang, Y.; Zhang, Z.; Hou, J.; He, D.; Xie, M.; Xu, L.; De, W.; Wang, Z.; et al. Lncrna hoxa11-as promotes proliferation and invasion of gastric cancer by scaffolding the chromatin modification factors prc2, lsd1, and dnmt1. Cancer Res. 2016, 76, 6299–6310. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Y.; Zhou, M.; Zhang, Y.; Wang, P.; Li, X.; Yang, J.; Wang, H.; Ding, Z. Hoxa9 inhibits hif-1alpha-mediated glycolysis through interacting with crip2 to repress cutaneous squamous cell carcinoma development. Nat. Commun. 2018, 9, 1480. [Google Scholar] [CrossRef]
- Jiang, Y.; Yan, B.; Lai, W.; Shi, Y.; Xiao, D.; Jia, J.; Liu, S.; Li, H.; Lu, J.; Li, Z.; et al. Repression of hox genes by lmp1 in nasopharyngeal carcinoma and modulation of glycolytic pathway genes by hoxc8. Oncogene 2015, 34, 6079–6091. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, metabolism, and cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Jogi, A.; Vaapil, M.; Johansson, M.; Pahlman, S. Cancer cell differentiation heterogeneity and aggressive behavior in solid tumors. Ups. J. Med. Sci. 2012, 117, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic inactivation of a ras association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 2000, 25, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the warburg effect: The role of hif-1 and pi3k. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Progression | HOX Gene | Tumor Cells Type | Function |
|---|---|---|---|
| Angiogenesis | HOXB7 [111,112,113] | Breast cancer Multiple myeloma | Upregulated HOXB7 drives angiogenic gene expression |
| HOXB9 [114,115] | Ovarian cancer Renal cancer Breast cancer | Downregulated HOXB9 attenuates angiogenic gene expression | |
| HOXA11-AS [116] | NSCLC | Upregulated HOXA11-AS promotes angiogenesis | |
| HOXA5 [117,118] | ECs | Sustained HOXA5 expression downregulates angiogenic genes and upregulates anti-angiogenic genes | |
| Autophagy | HOXC9 [119,120] | Glioblastoma | HOXC9 acts as a transcription inhibitor to directly binding to the promoter of DAPK1 |
| HOXC6 [121] | NPC | Downregulated HOXC6 promotes apoptosis and autophagy by inhibiting the TGF-β1/mTOR pathway | |
| HOTAIR [122,123] | Cervical cancer; Breast cancer; Chondrosarcoma | Downregulated HOTAIR inhibits autophagy | |
| Differentiation | HOXA clusters (except HOXA2 and HOXA5) [124] | Hematopoietic cells | HOXA genes except HOXA2 and HOXA5 induce delayed hematopoietic differentiation |
| HOXA9 [98,125,126,127] | Hematopoietic and lymphoid cancer. | HOXA9 involves in blocking differentiation NUP98–HOXA9 fusion, cooperation of HOXA9 with either Meis1 or FOXC1 inhibit differentiation | |
| HOXA10 [128,129] | Prostate cancer; OEA | HOXA10 blocks or promotes differentiation in a cancer-type-dependent manner | |
| HOXB8 [130] | HL-60 cells | HOXB8 blocks DMSO-induced granulocytic differentiation | |
| HOXC9 [131] | Neuroblastoma | HOXC9 promotes neuronal differentiation | |
| HOXA5 [132,133] | Colon cancer | Upregulated HOXA5 promotes differentiation of cancer stem cells | |
| HOTAIR [134] | Urothelial carcinoma | HOTAIR overexpression may affect differentiation state | |
| Apoptosis | HOXA5 [135,136,137,138,139,140,141,142] | Breast cancer; Leukemia; Osteosarcoma; Lung and cervical cancer | HOXA5 could activate apoptosis by upregulating p53 expression or activating caspase 2 and caspase 8; HOXA5 is involved in RA-mediated apoptosis |
| HOXA10 [143] | Breast cancer | HOXA10 could activate apoptosis by upregulating p53 expression | |
| HOXC6 [144,145,146] | HNSCC; Cervical cancer; Prostate cancer | HOXC6 plays an important anti-apoptotic role through regulating the expression of bcl-2 or suppressing NEP/MME and IGFBP-3 genes | |
| HOXA9 [126,147] | Leukemia | HOXA9 functions as an apoptosis suppressor by cooperating with JAK3/STAT5; HOXA9 could eliminate Meis1a-mediated apoptosis | |
| Proliferation | HOXA1 [148,149] | Breast cancer | HOXA1 promotes cell proliferation and survival by activating p44/42 MAPK signaling pathway or NF-κB pathway; |
| HOXA9 [150] | Leukemia | HOXA9 upregulates Igf1 to promote proliferation and survival | |
| HOXB7 [151,152] | Colorectal cancer Hepatocellular carcinoma | HOXB7 promotes cell proliferation and growth by accelerating G1-S transitions | |
| HOXC6 [153] | Gastrointestinal carcinoids cells | HOXC6 promotes cell proliferation by activating the oncogenic AP-1 signaling pathway | |
| HOXB3 [154] | NCI-H1437 cells A549 cells | HOXB3 promotes cell proliferation through silencing gene RASSFA1 | |
| HOXD3 [155] | Hepatocellular carcinoma | HOXD3 promotes proliferation and anti-apoptosis by activating MAPK/AKT cell signaling pathways | |
| HOXB9 [156] | HL cell lines | HOXB9 upregulated by ERK5 signal promotes proliferation and anti-apoptosis | |
| HOXC5 [157,158] | Thymoma; TGCT | HOXC5 inhibits proliferation by inhibiting hTERT expression | |
| HOXA10 [159,160] | Myeloid leukemia; Testicular cancer | Overexpressed HOXA10 stimulates the proliferation in myeloid leukemia; HOXA10 also inhibits cell proliferation during G2/M phases in testicular cancer cells | |
| Invasion and Metastasis | HOXA9 [161] | Breast cancer cell | HOXA9 expression could reduce bone metastasis |
| HOXA10 [162] | Endometrial carcinoma | HOXA10 suppresses invasion by inhibiting EMT | |
| HOXB1 and HOXB3 [163] | Pancreatic cancer | HOXB1 and HOXB3 downregulation facilitates invasion and metastasis | |
| HOXD10 [156,164,165,166,167] | Breast cancer | HOXD10 downregulation suppresses invasion | |
| HOXB7 [168,169,170] | Breast cancer | HOXB7 overexpression induces invasive and metastatic by activating the TGFβ signaling pathway | |
| HOXA11-AS [171] | Gastric cancer | HOXA11-AS expression promotes metastasis and invasion | |
| Metabolism | HOXA9 [172] | cSCC | HOXA9 inhibits glycolysis by negatively regulating HIF-1α |
| HOXC8 [173] | Nasopharyngeal carcinoma | HOXC8 downregulates glycolysis-related genes and upregulates TCA cycle-related genes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Huang, Q.; Wei, G.-H. The Role of HOX Transcription Factors in Cancer Predisposition and Progression. Cancers 2019, 11, 528. https://doi.org/10.3390/cancers11040528
Li B, Huang Q, Wei G-H. The Role of HOX Transcription Factors in Cancer Predisposition and Progression. Cancers. 2019; 11(4):528. https://doi.org/10.3390/cancers11040528
Chicago/Turabian StyleLi, Bo, Qilai Huang, and Gong-Hong Wei. 2019. "The Role of HOX Transcription Factors in Cancer Predisposition and Progression" Cancers 11, no. 4: 528. https://doi.org/10.3390/cancers11040528
APA StyleLi, B., Huang, Q., & Wei, G.-H. (2019). The Role of HOX Transcription Factors in Cancer Predisposition and Progression. Cancers, 11(4), 528. https://doi.org/10.3390/cancers11040528