NMDA Receptor-Mediated Signaling Pathways Enhance Radiation Resistance, Survival and Migration in Glioblastoma Cells—A Potential Target for Adjuvant Radiotherapy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
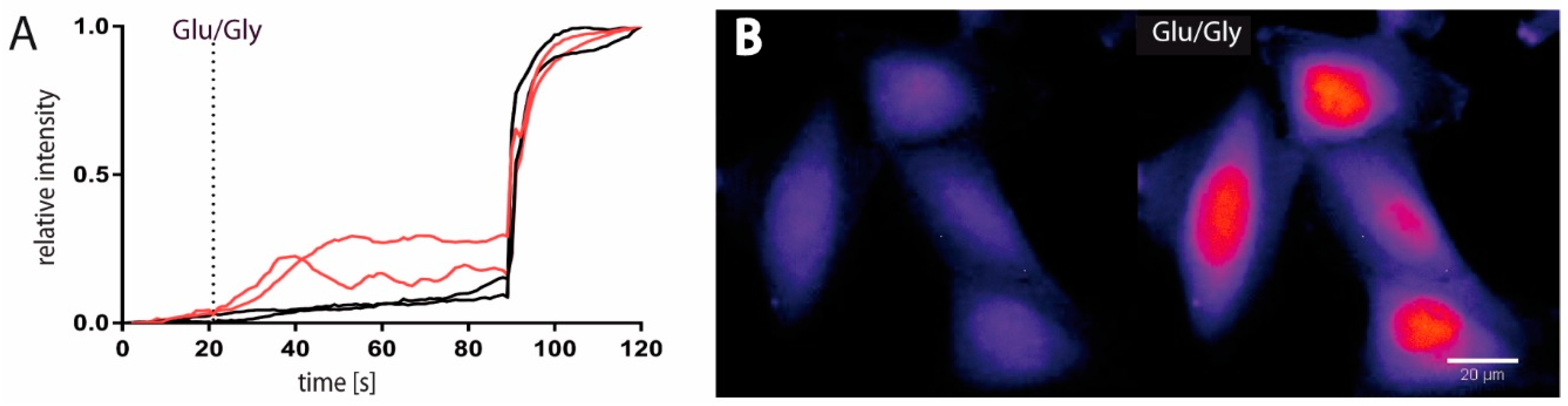
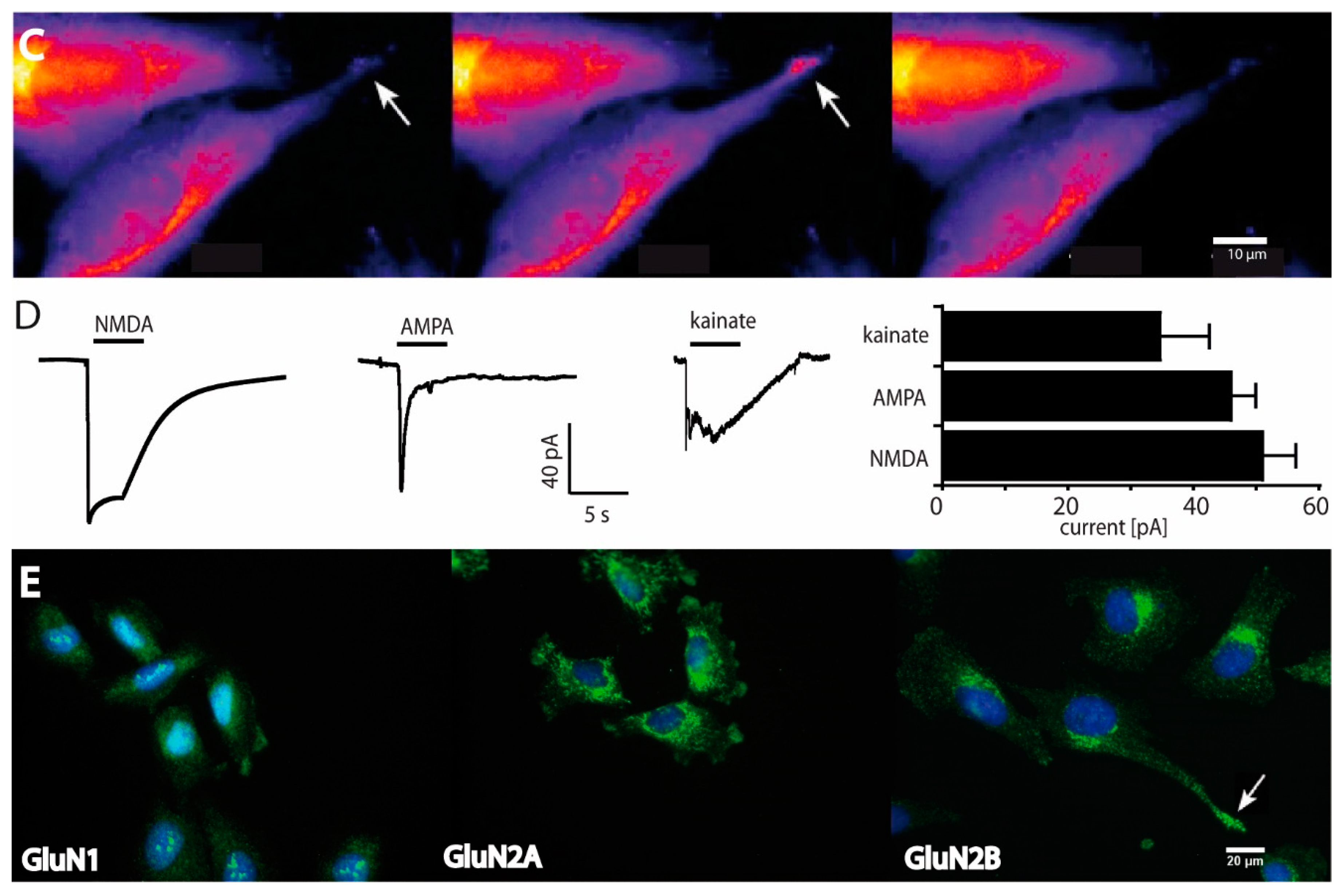
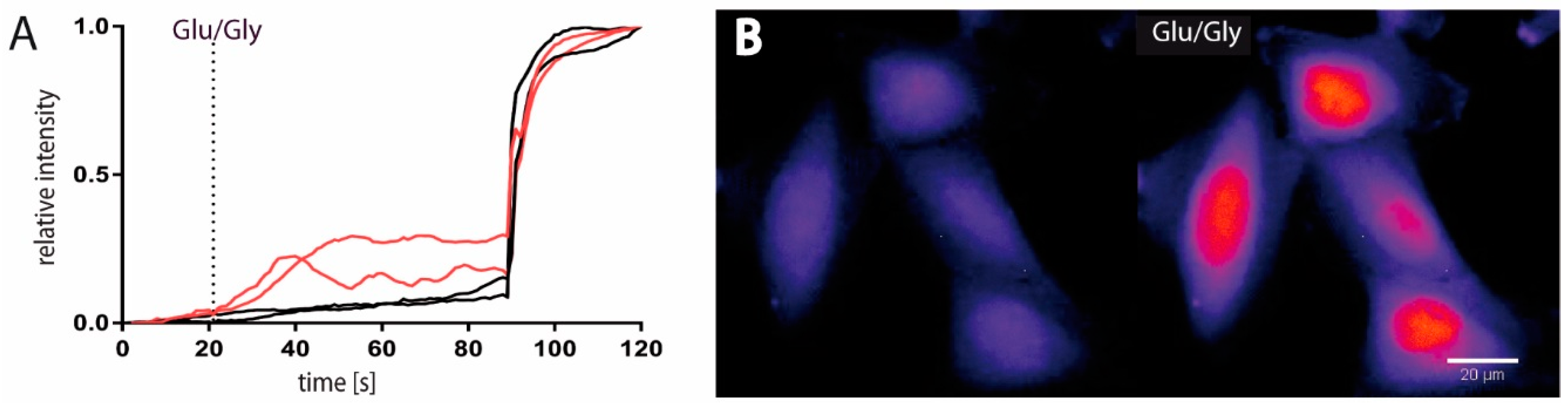
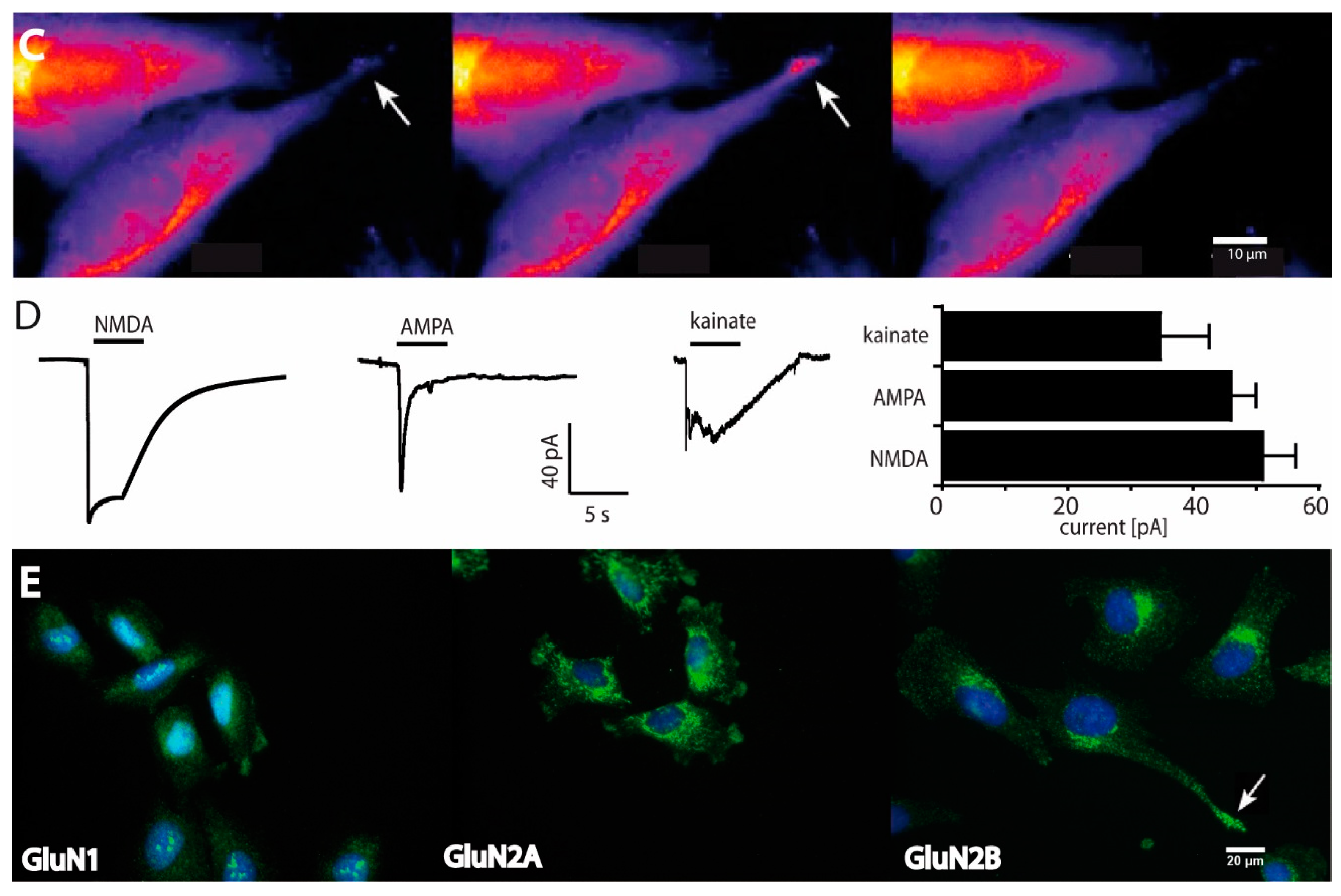
2.1. Functional and Immunohistochemical Characterization of NMDARs in LN229 Glioblastoma Cells
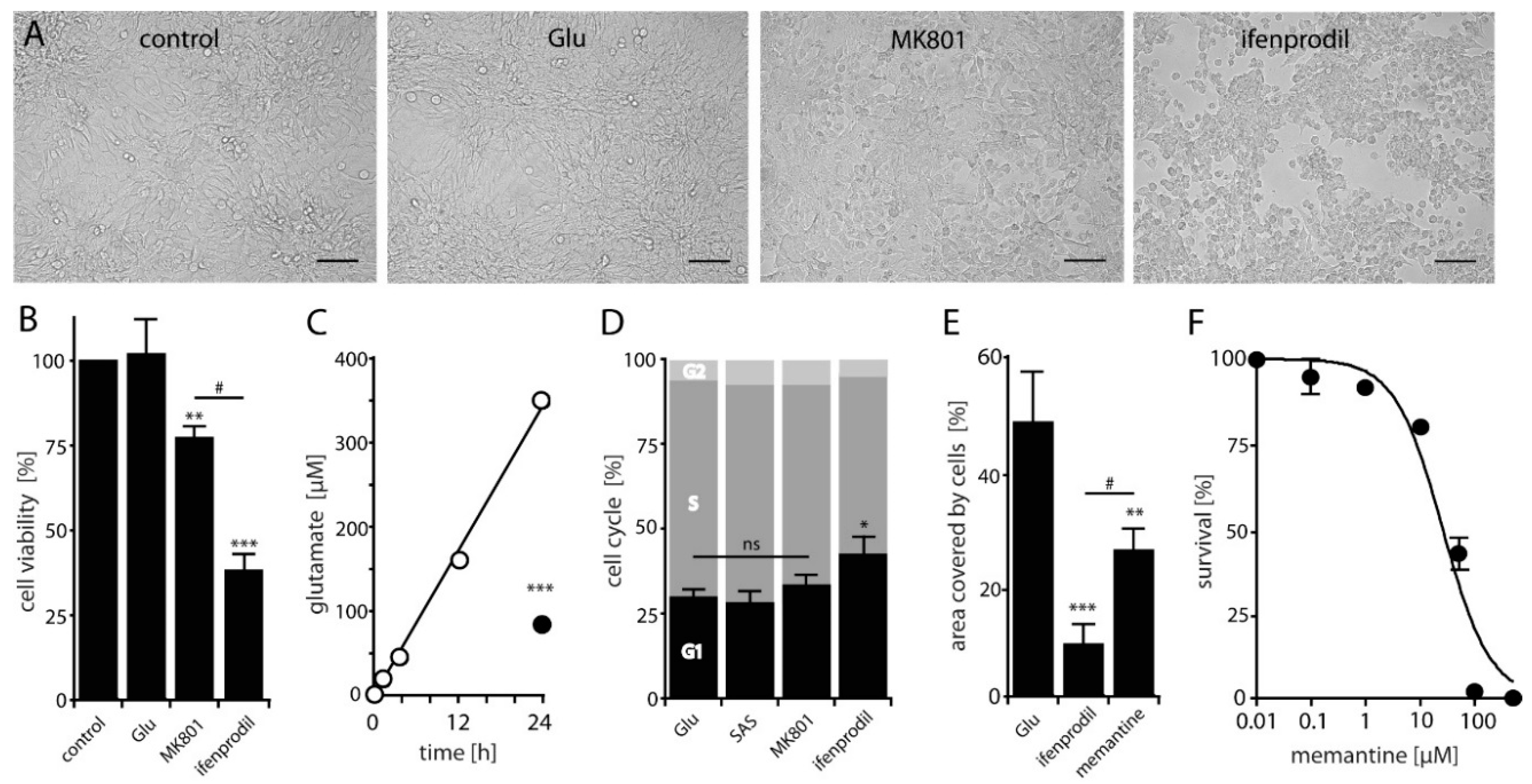
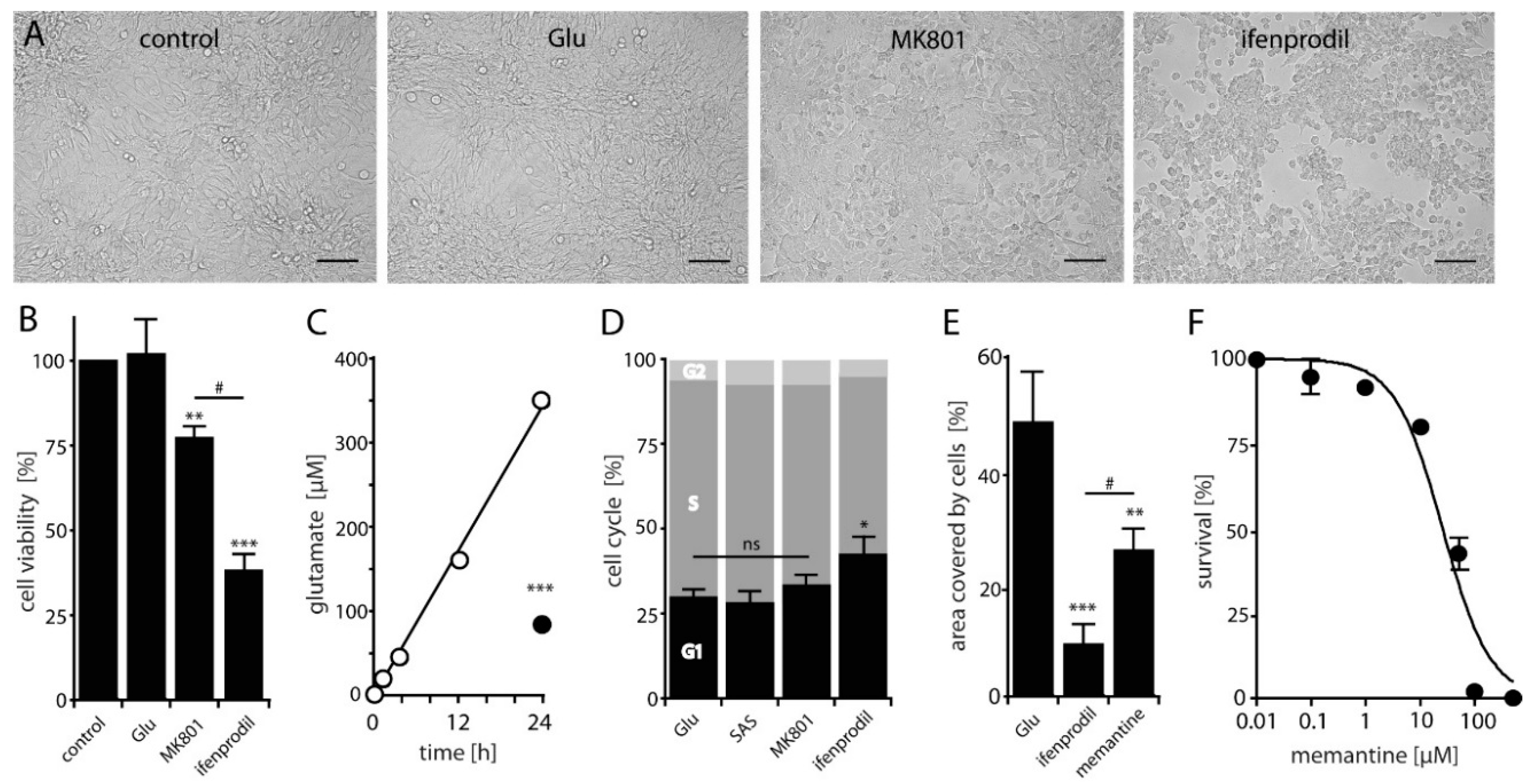
2.2. NMDAR-Activation is Crucial for LN229 Cell Viability, Migration and Survival
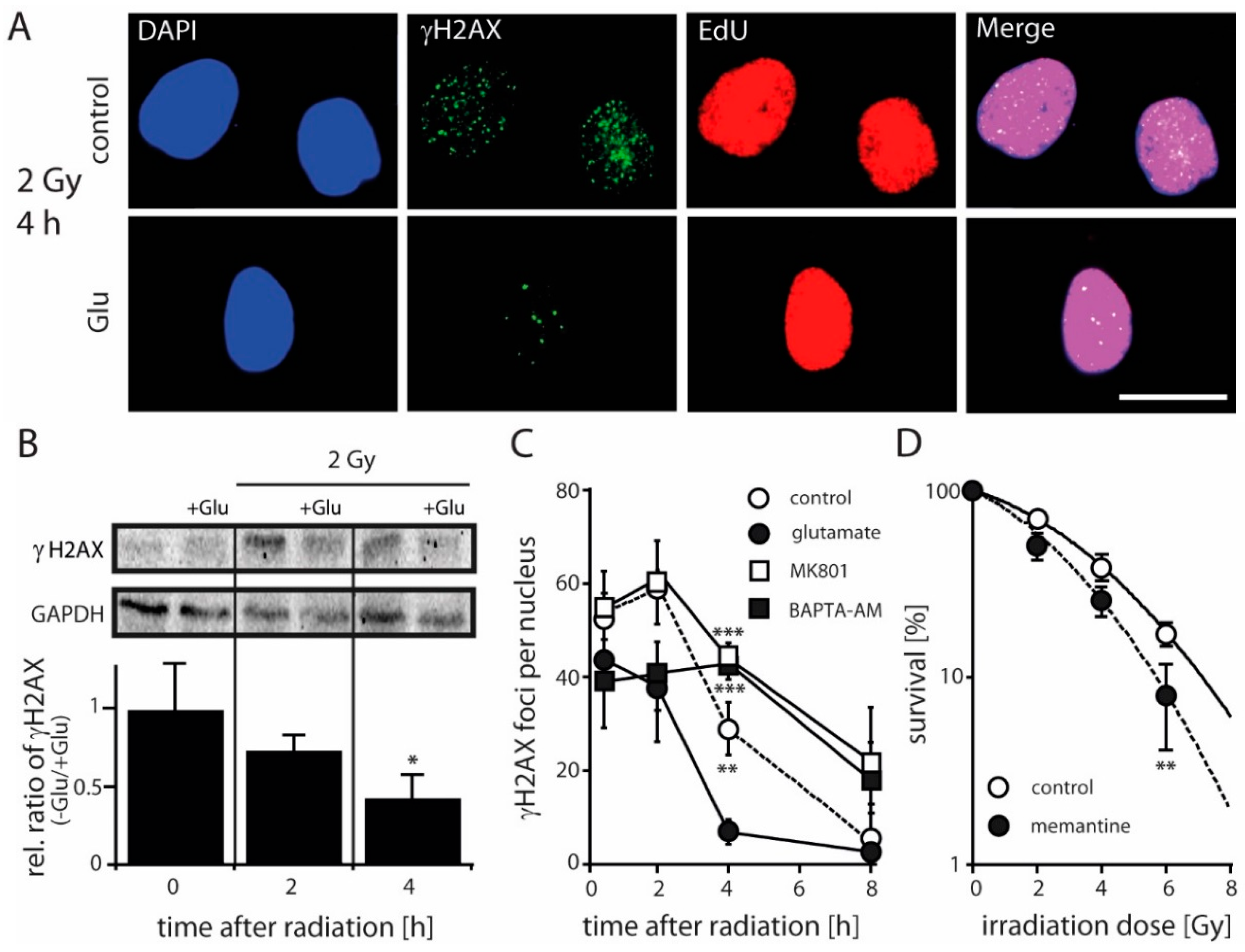
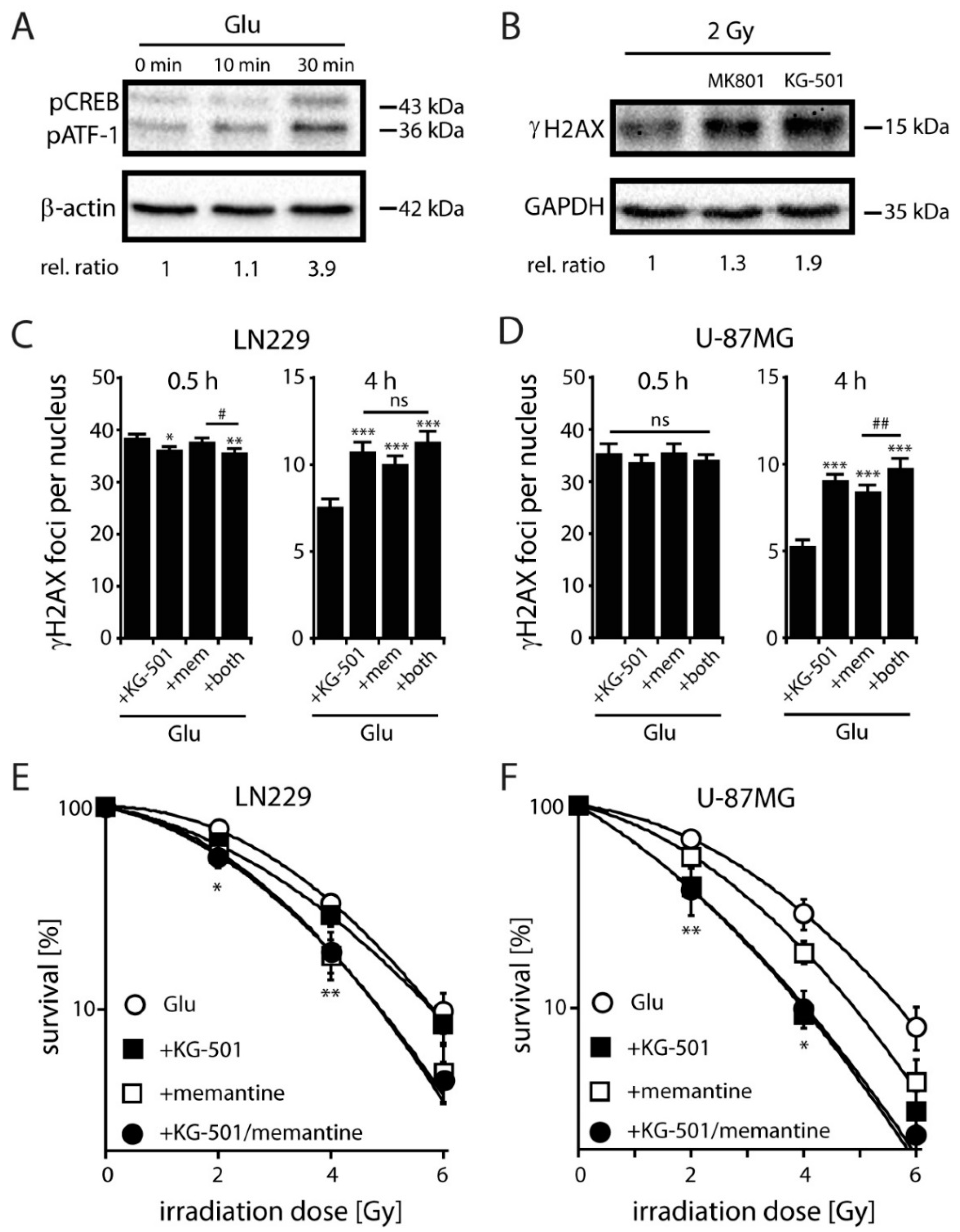
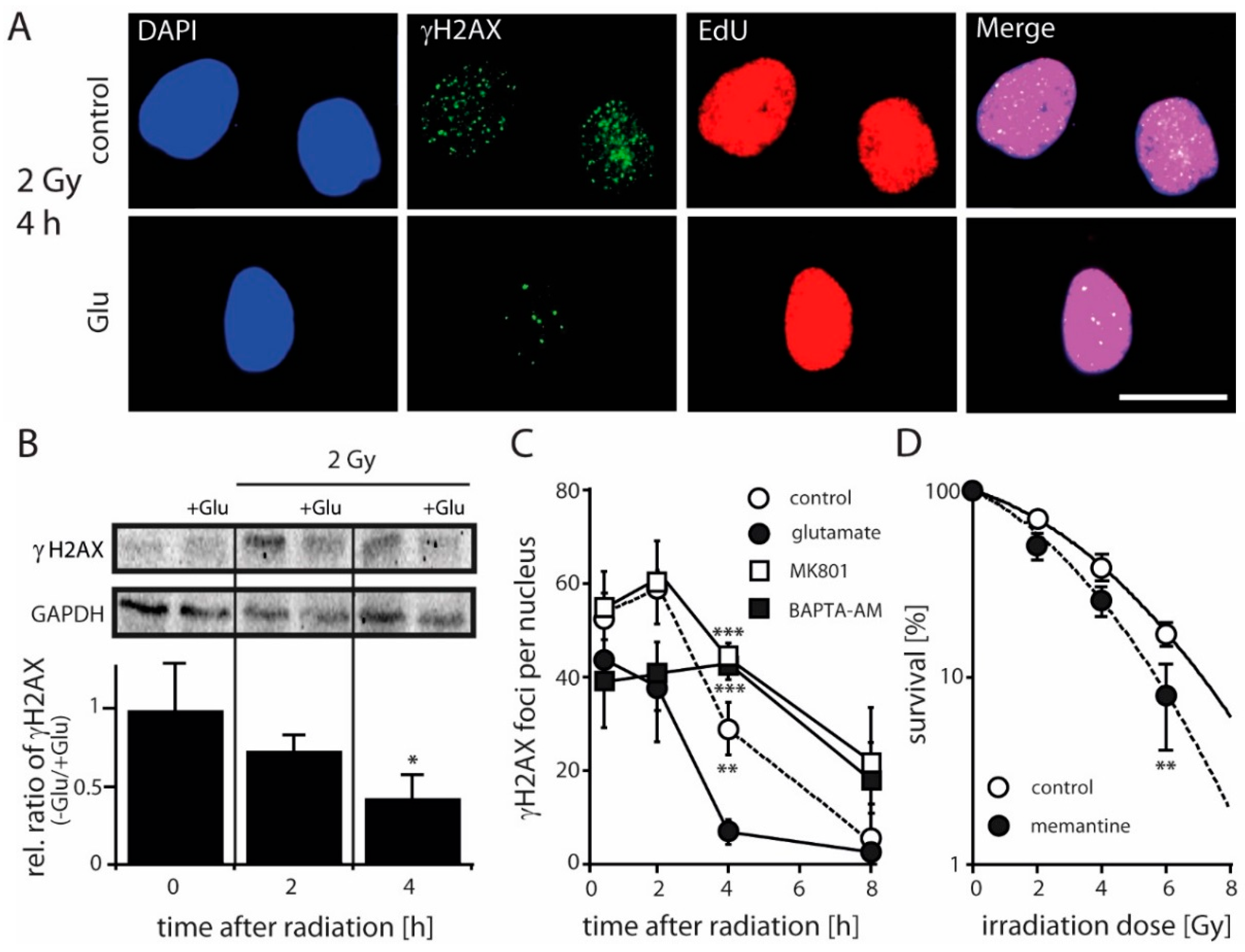
2.3. Antagonizing NMDARs Increases LN229 Radiosensitivity and Impairs Radiation-Induced DNA Double-Strand Break Repair
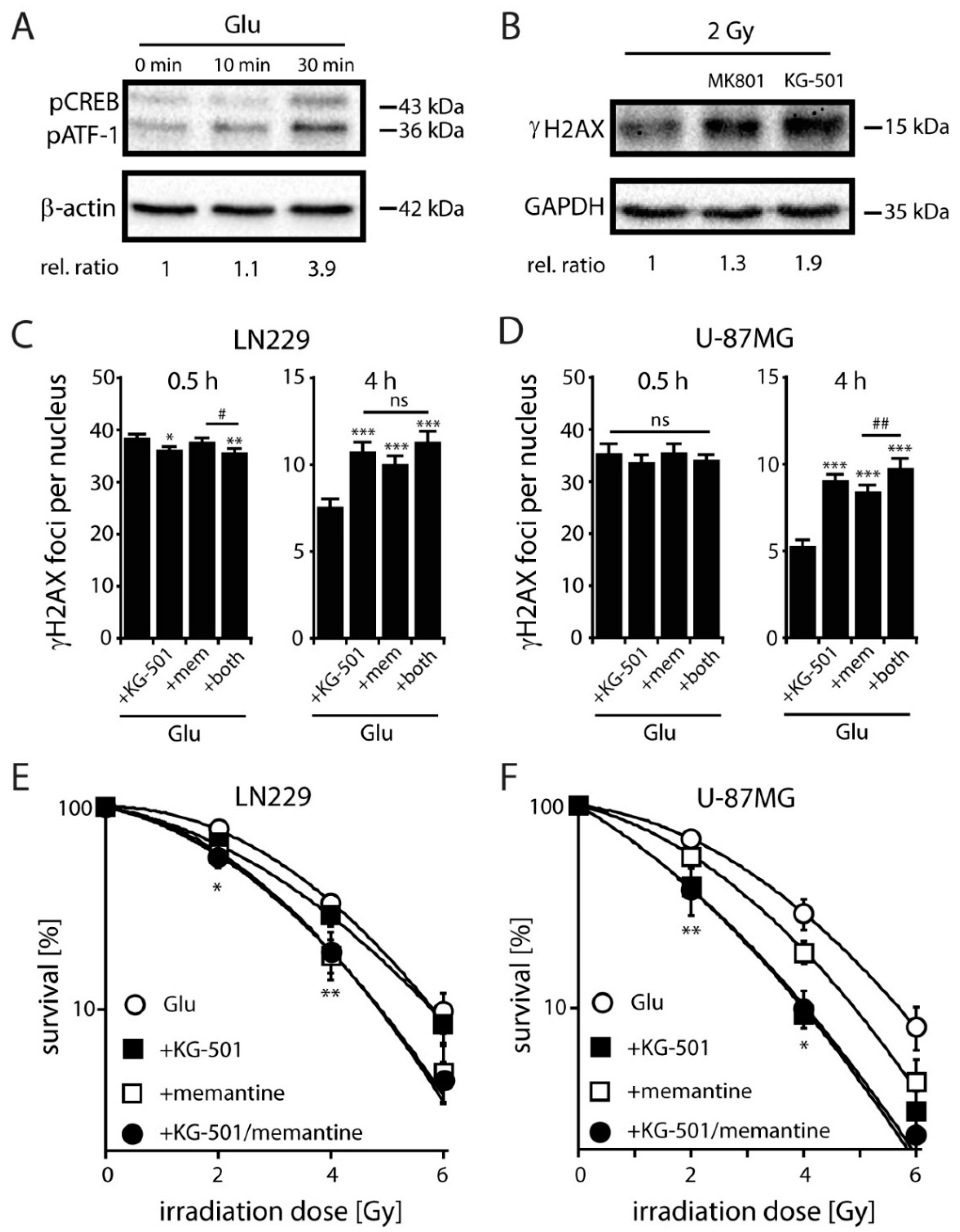
2.4. NMDARs Increase DSB Repair Capacity and Clonogenic Survival in LN229 and U-87MG Cells by Activation of the Transcription Factor CREB
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemical Treatment and X-Irradiation
4.3. Measurement of Extracellular Glutamate and BDNF Concentrations
4.4. 3-(4,5-Methylthiazol-2-yl)-2,5-Diphenyl-Tetrazolium Bromide (MTT) Assay
4.5. Clonogenic Survival Assay
4.6. Western Blot Analysis
4.7. Immunofluorescence Staining
4.8. γH2AX/EdU Double-Staining
4.9. Fluorescence-Activated Cell Cycle Analysis
4.10. Migration Assay
4.11. Electrophysiology
4.12. Calcium Imaging
4.13. Data Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 215–225. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, Y.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Atkins, R.J.; Ng, W.; Stylli, S.S.; Hovens, C.M.; Kaye, A.H. Repair mechanisms help glioblastoma resist treatment. J. Clin. Neurosci. 2015, 22, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Manini, I.; Caponnetto, F.; Bartolini, A.; Ius, T.; Mariuzzi, L.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D. Role of Microenvironment in Glioma Invasion: What We Learned from In Vitro Models. Int. J. Mol. Sci. 2018, 19, 147. [Google Scholar] [CrossRef] [PubMed]
- Marcus, H.J.; Carpenter, K.L.; Price, S.J.; Hutchinson, P.J. In vivo assessment of high-grade glioma biochemistry using microdialysis: A study of energy-related molecules, growth factors and cytokines. J. Neurooncol. 2010, 97, 11–23. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.; Sontheimer, H. Glutamate and the biology of gliomas. Glia 2011, 59, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Rothstein, J.D.; Sontheimer, H. Compromised glutamate transport in human glioma cells: Reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J. Neurosci. 1999, 19, 10767–10777. [Google Scholar] [CrossRef]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; von Lehe, M. Glioma-related seizures: Glutamate is the key. Nat. Med. 2011, 17, 1190. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The x(c)-cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell. Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.J.; Lyons, S.A.; Nelson, G.M.; Hamza, H.; Gladson, C.L.; Gillespie, G.Y.; Sontheimer, H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 2005, 25, 7101–7110. [Google Scholar] [CrossRef]
- Huberfeld, G.; Vecht, C.J. Seizures and gliomas—towards a single therapeutic approach. Nat. Rev. Neurol. 2016, 12, 204–216. [Google Scholar] [CrossRef]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar] [PubMed]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vöckler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef]
- Prickett, T.D.; Samuels, Y. Molecular pathways: Dysregulated glutamatergic signaling pathways in cancer. Clin. Cancer Res. 2012, 18, 4240–4246. [Google Scholar] [CrossRef]
- Bozic, M.; Valdivielso, J.M. The potential of targeting NMDA receptors outside the CNS. Expert Opin. Ther. Targets 2015, 19, 399–413. [Google Scholar] [CrossRef]
- Ribeiro, M.P.; Custodio, J.B.; Santos, A.E. Ionotropic glutamate receptor antagonists and cancer therapy: Time to think out of the box? Cancer Chemother. Pharmacol. 2017, 79, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Yamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Tamura, M.; et al. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar] [CrossRef]
- Rzeski, W.; Turski, L.; Ikonomidou, C. Glutamate antagonists limit tumor growth. Proc. Natl. Acad. Sci. USA 2001, 98, 6372–6377. [Google Scholar] [CrossRef]
- de Groot, J.F.; Piao, Y.; Lu, L.; Fuller, G.N.; Yung, W.A. Knockdown of GluR1 expression by RNA interference inhibits glioma proliferation. J. Neurooncol. 2008, 88, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Stepulak, A.; Luksch, H.; Gebhardt, C.; Uckermann, O.; Marzahn, J.; Sifringer, M.; Rzeski, W.; Staufner, C.; Brocke, K.S.; Turski, L.; et al. Expression of glutamate receptor subunits in human cancers. Histochem. Cell Biol. 2009, 132, 435–445. [Google Scholar] [CrossRef]
- Noch, E.; Khalili, K. Molecular mechanisms of necrosis in glioblastoma: The role of glutamate excitotoxicity. Cancer Biol. Ther. 2009, 8, 1791–1797. [Google Scholar] [CrossRef]
- Goudar, R.K.; Keir, S.T.; Bigner, D.D.; Friedman, H.S. NMDA and AMPA glutamate receptor antagonists in the treatment of human malignant glioma xenografts. Exp. Mol. Ther. 2004, 64, 41. [Google Scholar]
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007, 67, 9463–9471. [Google Scholar] [CrossRef]
- Li, L.; Hanahan, D. Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 2013, 153, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, P.; Devi, N.A.; Fathima, K.H.; Nanjaiah, N. Activation of NMDA receptor of glutamate influences MMP-2 activity and proliferation of glioma cells. Neurol. Sci. 2014, 35, 823–829. [Google Scholar] [CrossRef]
- Takano, T.; Lin, J.H.C.; Arcuino, G.; Gao, Q.; Yang, J.; Nedergaard, M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001, 7, 1010–1015. [Google Scholar] [CrossRef]
- Laube, B.; Kuhse, J.; Betz, H. Evidence for a tetrameric structure of recombinant NMDA receptors. J. Neurosci. 1998, 18, 2954–2961. [Google Scholar] [CrossRef] [PubMed]
- Laube, B.; Hirai, H.; Sturgess, M.; Betz, H.; Kuhse, J. Molecular determinants of agonist discrimination by NMDA receptor subunits: Analysis of the glutamate binding site on the NR2B subunit. Neuron 1997, 18, 493–503. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Deutsch, S.I.; Tang, A.H.; Burket, J.A.; Benson, A.D. NMDA receptors on the surface of cancer cells: Target for chemotherapy? Biomed. Pharmacother. 2014, 68, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Stepulak, A.; Rola, R.; Polberg, K.; Ikonomidou, C. Glutamate and its receptors in cancer. J. Neural Transm. (Vienna) 2014, 121, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, A.; Koiri, K.R. N-Methyl-D-Aspartate (NMDA) Receptors: Therapeutic Target against Cancer. Int. J. Immunother. Cancer Res. 2015, 1, 17. [Google Scholar]
- Van Vuurden, D.G.; Yazdani, M.; Bosma, I.; Broekhuizen, A.J.; Postma, T.J.; Heimans, J.J.; van der Valk, P.; Aronica, E.; Tannous, B.A.; Würdinger, T.; et al. Attenuated AMPA receptor expression allows glioblastoma cell survival in glutamate-rich environment. PLoS ONE 2009, 4, e5953. [Google Scholar] [CrossRef]
- D’Auria, F.; Centurione, L.; Centurione, M.A.; Angelini, A.; Di Pietro, R. Regulation of Cancer Cell Responsiveness to Ionizing Radiation Treatment by Cyclic AMP Response Element Binding Nuclear Transcription Factor. Front. Oncol. 2017, 7, 76. [Google Scholar] [CrossRef]
- Best, J.L.; Amezcua, C.A.; Mayr, B.; Flechner, L.; Murawsky, C.M.; Emerson, B.; Zor, T.; Gardner, K.H.; Montminy, M. Identification of small-molecule antagonists that inhibit an activator: Coactivator interaction. Proc. Natl. Acad. Sci. USA 2004, 101, 17622–17627. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Heckel, A.; Hahnen, E.; Engelhorn, T.; Doerfler, A.; Ganslandt, O.; Nimsky, C.; Buchfelder, M.; Eyüpogl, I.Y. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 2008, 14, 629–632. [Google Scholar] [CrossRef] [PubMed]
- North, W.G.; Gao, G.; Memoli, V.A.; Pang, R.H.; Lynch, L. Breast cancer expresses functional NMDA receptors. Breast Cancer Res. Treat. 2010, 122, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Lefranc, F.; Le Rhun, E.; Kiss, R.; Weller, M. Glioblastoma quo vadis: Will migration and invasiveness reemerge as therapeutic targets? Cancer Treat. Rev. 2018, 68, 145–154. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Kastan, M.B. Cell cycle control and cancer. Science 1994, 266, 1821–1828. [Google Scholar] [CrossRef]
- Cho, E.C.; Mitton, B.; Sakamoto, K.M. CREB and leukemogenesis. Crit. Rev. Oncog. 2011, 16, 37–46. [Google Scholar] [CrossRef]
- Yang, Y.M.; Dolan, L.R.; Ronai, Z. Expression of dominant negative CREB reduces resistance to radiation of human melanoma cells. Oncogene 1996, 12, 2223–2233. [Google Scholar]
- Xie, S.; Price, J.E.; Luca, M.; Jean, D.; Ronai, Z.; Bar-Eli, M. Dominant-negative CREB inhibits tumor growth and metastasis of human melanoma cells. Oncogene 1997, 15, 2069–2075. [Google Scholar] [CrossRef]
- Amorino, G.P.; Mikkelsen, R.B.; Valerie, K.; Schmidt-Ullrich, R.K. Dominant-negative cAMP-responsive element-binding protein inhibits proliferating cell nuclear antigen and DNA repair, leading to increased cellular radiosensitivity. J. Biol. Chem. 2003, 278, 29394–29399. [Google Scholar] [CrossRef]
- Schmidt, R.H.; Nickerson, J.M.; Boatright, J.H. Exercise as Gene Therapy: BDNF and DNA Damage Repair. Asia Pac. J. Ophthalmol. 2016, 5, 309–311. [Google Scholar] [CrossRef]
- Xiong, J.; Zhou, L.; Lim, Y.; Yang, M.; Zhu, Y.H.; Li, Z.W.; Zhou, F.H.; Xiao, Z.C.; Zhou, X.F. Mature BDNF promotes the growth of glioma cells in vitro. Oncol. Rep. 2013, 30, 2719–2724. [Google Scholar] [CrossRef]
- Yano, S.; Tokumitsu, H.; Soderling, T.R. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 1998, 396, 584–587. [Google Scholar] [CrossRef]
- Lutz, H.; Nguyen, T.; Joswig, J.; Rau, K.; Laube, B. NMDA receptor signaling mediates cFos expression via Top2β-induced DSBs in Glioblastoma cells. Cancers 2019, 11, 306. [Google Scholar] [CrossRef]
- Laube, B.; Kuhse, J.; Betz, H. Kinetic and mutational analysis of Zn2+ modulation of recombinant human inhibitory glycine receptors. J. Physiol. 2000, 522, 215–230. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller-Längle, A.; Lutz, H.; Hehlgans, S.; Rödel, F.; Rau, K.; Laube, B. NMDA Receptor-Mediated Signaling Pathways Enhance Radiation Resistance, Survival and Migration in Glioblastoma Cells—A Potential Target for Adjuvant Radiotherapy. Cancers 2019, 11, 503. https://doi.org/10.3390/cancers11040503
Müller-Längle A, Lutz H, Hehlgans S, Rödel F, Rau K, Laube B. NMDA Receptor-Mediated Signaling Pathways Enhance Radiation Resistance, Survival and Migration in Glioblastoma Cells—A Potential Target for Adjuvant Radiotherapy. Cancers. 2019; 11(4):503. https://doi.org/10.3390/cancers11040503
Chicago/Turabian StyleMüller-Längle, Adriana, Henrik Lutz, Stephanie Hehlgans, Franz Rödel, Kerstin Rau, and Bodo Laube. 2019. "NMDA Receptor-Mediated Signaling Pathways Enhance Radiation Resistance, Survival and Migration in Glioblastoma Cells—A Potential Target for Adjuvant Radiotherapy" Cancers 11, no. 4: 503. https://doi.org/10.3390/cancers11040503
APA StyleMüller-Längle, A., Lutz, H., Hehlgans, S., Rödel, F., Rau, K., & Laube, B. (2019). NMDA Receptor-Mediated Signaling Pathways Enhance Radiation Resistance, Survival and Migration in Glioblastoma Cells—A Potential Target for Adjuvant Radiotherapy. Cancers, 11(4), 503. https://doi.org/10.3390/cancers11040503