1. Introduction
Hydrogen peroxide (H
2O
2) plays a key role as a second messenger in cell signal transduction, regulating cell proliferation, differentiation, migration and apoptosis [
1,
2,
3]. This occurs primarily through the oxidation of redox-sensitive (reactive) cysteine (Cys) residues within proteins by H
2O
2 which results in changes to their structure and function [
4,
5,
6].
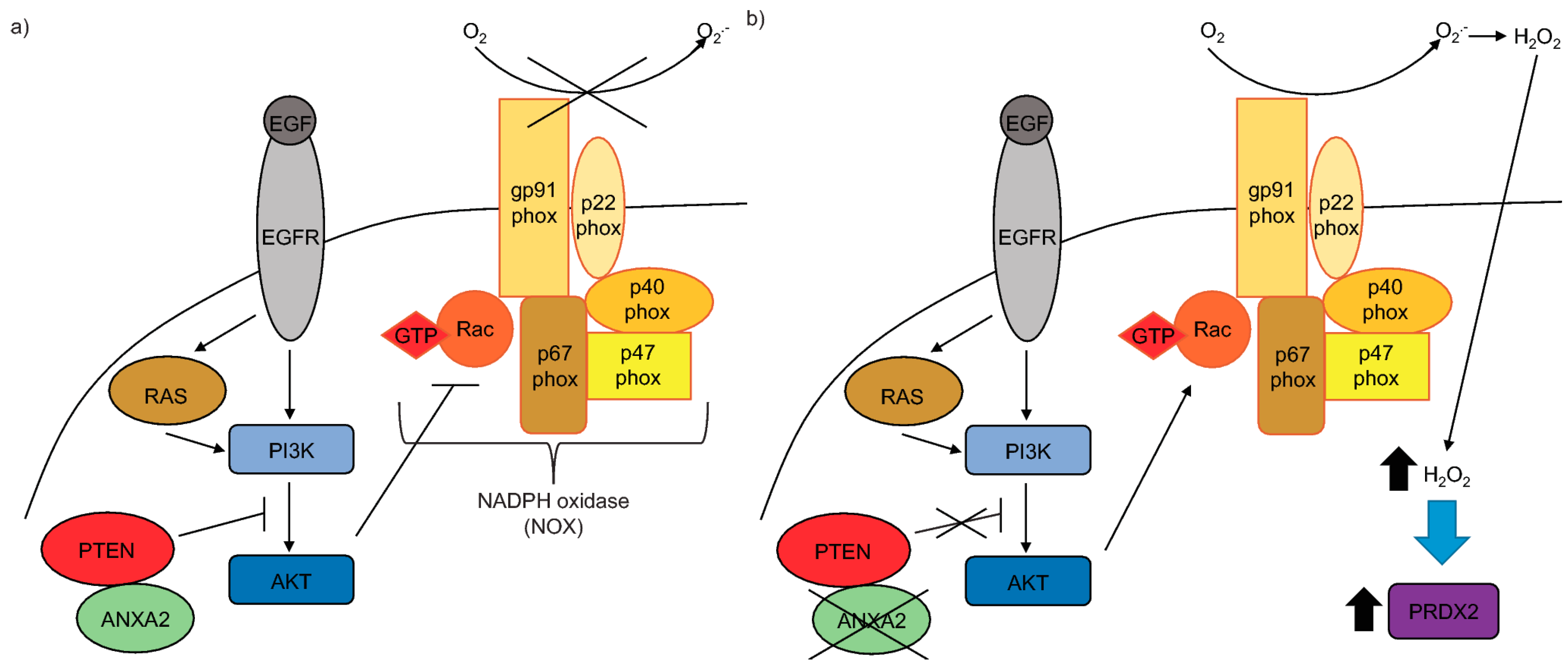
The epidermal growth factor receptor (EGFR) signaling is commonly up-regulated in cancer, constituting an important pathway that regulates cell growth, survival, proliferation and differentiation [
7,
8]. EGFR signaling is initiated by growth factor (e.g., EGF) binding which stimulates dimerization and autophosphorylation of the receptor and subsequent downstream signaling by phosphorylation/activation of tyrosine kinases and the concomitant inhibition of protein tyrosine phosphatases and the phosphatase and tensin homologue (PTEN) protein. Particularly, the activation of the EGFR/PI3K/AKT pathway enables Rac to bind to and activate NADPH oxidase (NOX), ultimately leading to the production of H
2O
2 [
9,
10]. H
2O
2 reversibly oxidizes and inactivates the main inhibitor of the PI3K/AKT pathway, PTEN, allowing signal transduction to occur.
Another prominent H
2O
2-dependent signaling network associated with tumorigenesis is the Ras pathway. Ras is a small G-coupled protein, whose activity is regulated by association with GTP (promoted by Guanine Exchange Factors) generating an ON/active protein or GDP (promoted by its own GTPase activity) constituting the OFF/inactive Ras. The constitutive activation of Ras has been shown in approximately 30% of all cancers [
11]. This occurs via mutation(s) of critical amino acid residue(s) leading to the loss of Ras GTPase activity [
12]. Ras regulates three critical signaling networks, namely the Raf/MEK/ERK, PI3K/AKT and Ral-GDS pathways [
12]. Ras/PI3K/AKT signaling induces ROS production through the activation of the NOX regulatory subunit, Rac [
13].
To balance the proliferative and survival advantage of moderate reactive oxygen species (ROS) levels versus their damaging effect at high concentrations, cancer cells undergo a redox adaptation process through the induction of the antioxidant response [
14]. We identified a novel redox regulatory protein, annexin A2 (ANXA2) that plays a key role in protecting cells from ROS induced death, supporting both tumor growth and chemo-resistance [
1,
4,
15]. ANXA2 can exist in the cells as a monomer or a heterotetramer, where two ANXA2 molecules are bound together by a dimer of the protein, S100A10 [
16,
17]. We previously demonstrated that the redox regulatory function of ANXA2 is independent of S100A10 [
1,
14,
15].
Here we show that ANXA2 depleted cells have enhanced phosphorylation of AKT upon EGF/EGFR activation or transformation with oncogenic H-RasV12. We demonstrate that ANXA2, via its reactive Cys-8 residue, can bind to the negative regulator of the PI3K/AKT signaling pathway, PTEN. Our co-expression studies reveal that ANXA2 positively regulates PTEN activity, leading to the inhibition of AKT. We also report for the first time that ANXA2 inversely regulates the peroxidase, PRDX2 in a reactive oxygen species (ROS) dependent manner.
3. Discussion
We previously identified ANXA2 as a redox regulatory protein that plays an important role during oxidative stress and tumorigenesis [
1]. With this work, we show for the first time, that depletion of ANXA2 in cancer cells leads to enhanced activation of AKT in response to either EGF/EGFR activation or oncogenic H-RasV12 transformation (
Figure 1 and
Supplementary Materials Figure S1). The main inhibitor of the PI3K/AKT pathway, PTEN, contains reactive Cys residues in its catalytic domain that can be readily oxidized by H
2O
2 inactivating its phosphatase function [
18]. Our co-immunoprecipitation studies revealed that ANXA2 interacts with PTEN via ANXA2 Cys-8 residue (
Figure 2A). These data in conjunction with the decreased activation of AKT observed in ANXA2-containing compared to ANXA2 depleted cells upon H
2O
2-dependent signaling (
Figure 1 and
Supplementary Materials Figure S1) suggested that ANXA2 positively regulates PTEN activity. Co-transfection studies using a combination of WT or mutants ANXA2 and PTEN expression plasmids, confirmed that ANXA2 positively regulates PTEN inhibition of pSer473 AKT, in a Cys-8 residue of ANXA2 dependent way (
Figure 2C,D). Importantly, expression of ANXA2 alone in 293T cells did not inhibit pSer473 AKT compared to 293T control cells (293T transfected with pcDNA3) (
Figure 2B, lane 3). These data suggest that ANXA2 does not directly regulate AKT activity, but does so via PTEN.
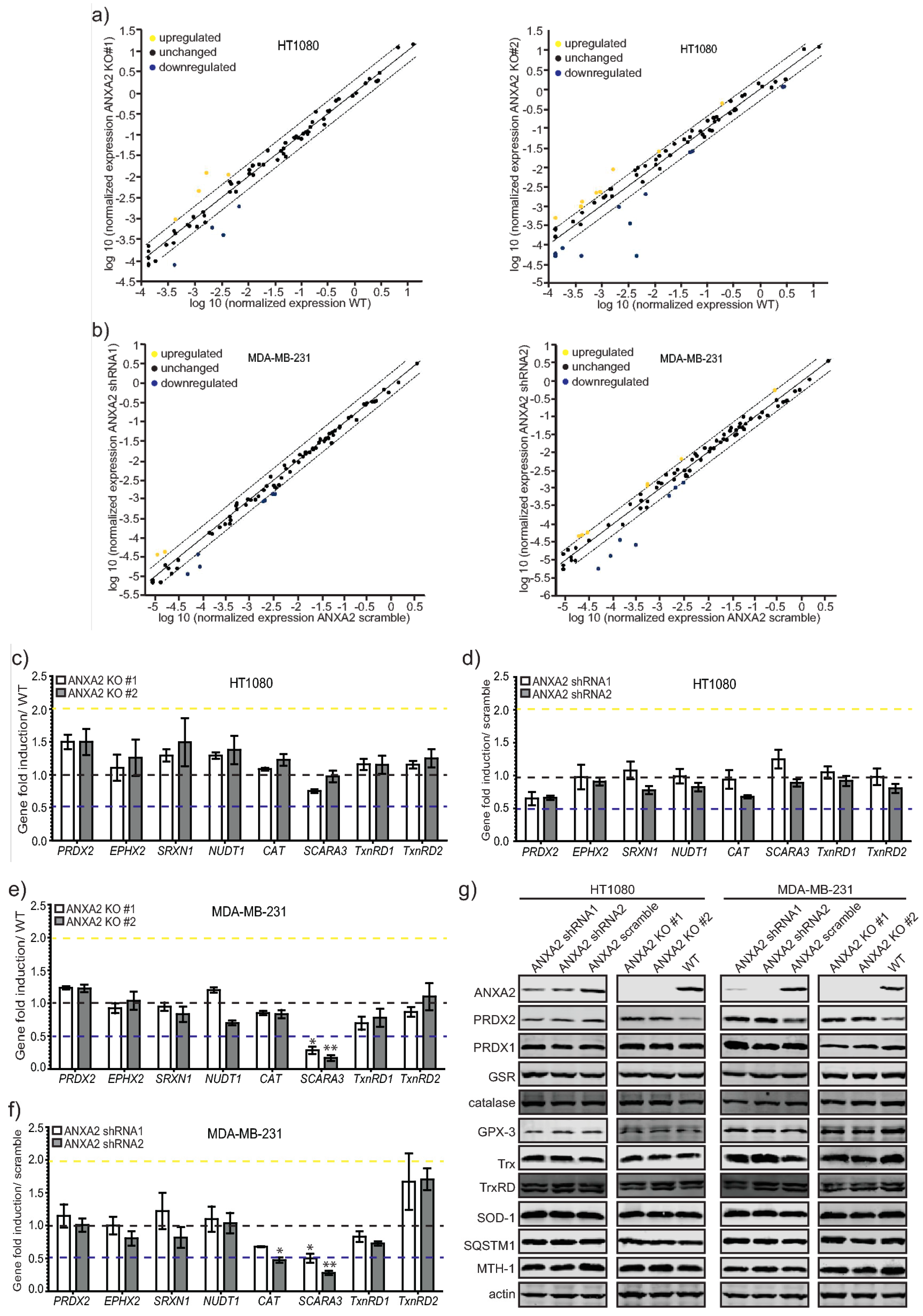
In this work we show for the first time that ANXA2 regulates the expression of the peroxidase, PRDX2. In summary, we observed increased expression of PRDX2 upon depletion of ANXA2 in cancer cells (
Figure 5G and
Supplementary Materials Figure S5). Of note, depletion of ANXA2 by shRNA was insufficient to induce PRDX2 up-regulation in HT1080 ANXA2 KD cells or in MDA-MB-231 and HT1080 pBABE cells, whose down-regulation of ANXA2 was less efficient compared to MDA-MB-231 ANXA2 KD cells (
Figures S3 and S5). However,
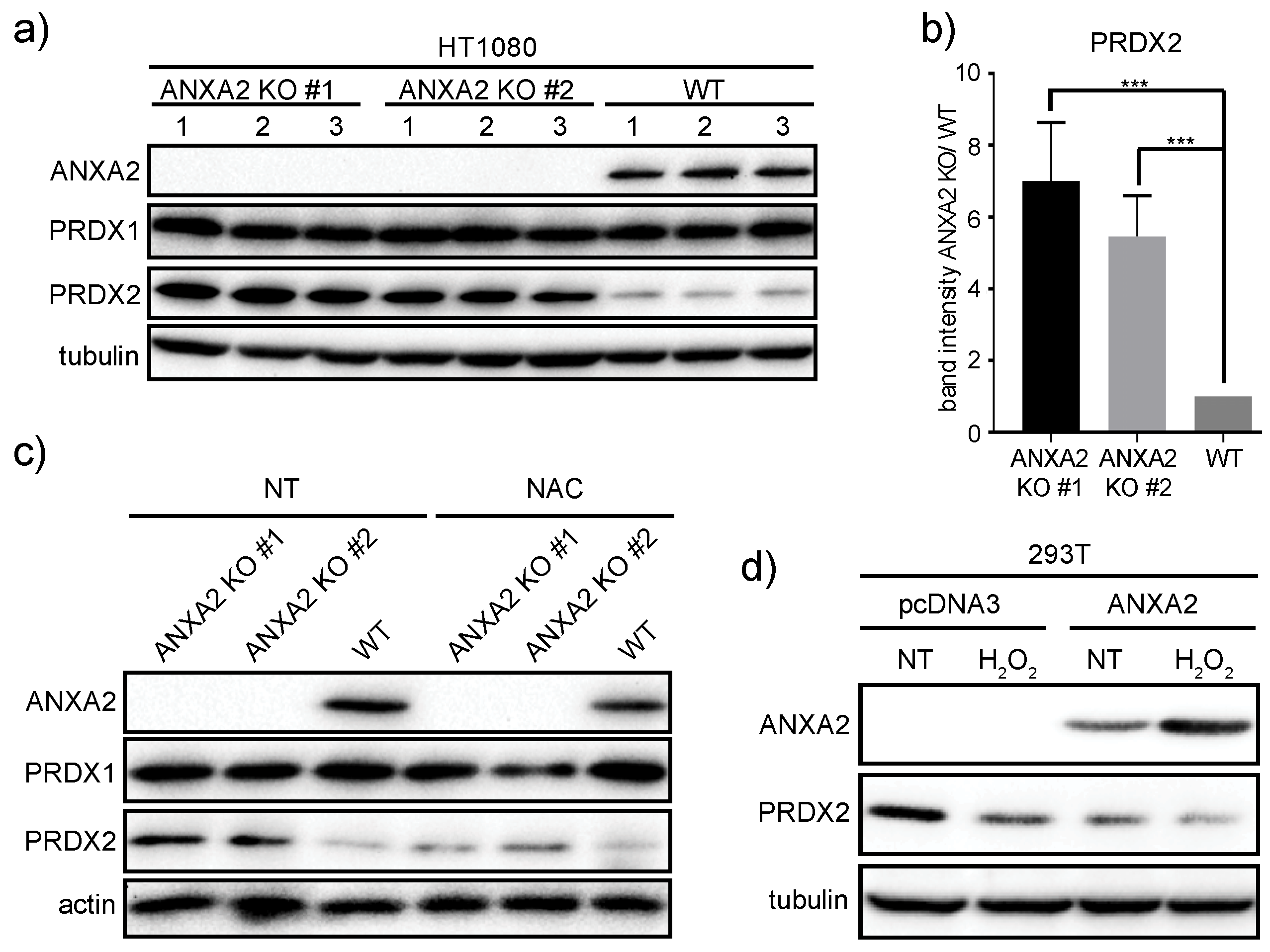
ANXA2 gene deletion in HT1080 and MDA-MB-231 cells led to the over-expression of PRDX2 compared to their respective WT cells (
Figure 3,
Figure 5 and
Figure S6). Together these results suggest that a threshold for ANXA2 depletion is required to trigger PRDX2 up-regulation in cancer cells. We demonstrated that PRDX2 regulation by ANXA2 was ROS dependent (
Figure 3C). In this sense, a pronounced down-regulation of ANXA2 might be necessary to induce sufficient accumulation of ROS leading to the subsequent up-regulation of PRDX2 to compensate for the REDOX imbalance. In fact, cells over-expressing H-RasV12, which leads to up-regulation of intracellular ROS, and depleted of ANXA2 showed a significantly higher expression of PRDX2 compared to cells expressing endogenous levels of ANXA2 and H-RasV12 (
Supplementary Materials Figure S5). Analysis of intracellular ROS revealed that HT1080 and A549 ANXA2 KD cells have significantly higher levels of endogenous ROS compared to their respective controls, while this was not observed in MDA-MB-231 ANXA2 KD cells (
Figure S4). We observed up-regulation of PRDX2 in the MDA-MB-231 ANXA2 KD compared to control cells, which did not occur in the isogenic HT1080 or A549 cells (
Supplementary Materials Figure S4). Taken together these data support that up-regulation of PRDX2 in MDA-MB-231 ANXA2 KD cells leads to REDOX homeostasis. Importantly, over-expression of ANXA2 led to down-regulation of PRDX2 in 293T cells (
Figure 3D), further supporting that ANXA2 inversely regulates the cellular expression of PRDX2. Interestingly, when we depleted PRDX2 in cancer cells we did not observe up-regulation of ANXA2 (
Supplementary Materials Figure S7). Analysis of ANXA2 and PRDX2 protein expression in a panel of cell lines further showed an inverse relationship between ANXA2 and PRDX2 levels (
Figure 4A). Furthermore, expression patterns of ANXA2 and PRDX2 in colon cancer sections revealed that the distribution of these two proteins is highly distinct. Areas with high ANXA2 expression display little to no PRDX2 and vice-versa (
Figure 4D and
Supplementary Materials Figure S8A). PRDX2 and ANXA2 share many characteristics, including similar cellular distribution, high cytoplasmic abundancy with a small fraction in the nucleus [
19,
20], calcium-dependent plasma membrane binding [
17,
21,
22], lipid raft co-localization [
20,
23], ion channels regulation [
17,
24,
25], both directly inactivate H
2O
2 and are recycled/reduced by the thioredoxin system [
1,
20] and both protect cellular DNA from oxidative damage [
15,
26]. However, it is highly likely that ANXA2 and PRDX2 also have distinct functions and interact with different proteins and targets within cells.
A study by Cao et al. found that PRDX1 binds to PTEN protecting it from oxidation-induced inactivation and indirectly regulating AKT phosphorylation upon Ras signaling [
27]. The authors also show that PRDX2 was not able to bind to PTEN, which is in agreement with our results, demonstrating that PRDX2 up-regulation is not able to impede the enhanced AKT phosphorylation observed upon depletion of ANXA2 in cancer cells.
Over-oxidation of PRDXs has been linked to cell signaling through the “floodgate model,” in which PRDXs consume low levels of endogenous peroxides, but enhanced generation leads to over-oxidation/degradation and increase in H
2O
2 that acts as a second messenger [
28]. Compared to PRDX2, ANXA2 seems to be more resistant to oxidation during H
2O
2-dependent signaling [
1,
4]. In fact, when we treated 293T cells with a high oxidative insult (100 µM of H
2O
2) we observed decreased levels of PRDX2, while ANXA2 expression increased (
Figure 3D). In these experiments we transfected 293T cells with the ANXA2-pcDNA3 plasmid to transiently express ANXA2 in these cells. It has been previously shown that H
2O
2 induces the CMV promoter [
29]. For this reason, it is likely that ANXA2 up-regulation in H
2O
2 treated cells is due at least in part to H
2O
2 induced CMV promoter activation. Nevertheless, Madureira et al. have previously shown that the treatment of ANXA2 low expressing cells, 293T and MCF7, with 100 μM H
2O
2 over a long period of time (two weeks) led to up-regulation of ANXA2 in these cells [
1].
4. Materials and Methods
4.1. Cell Culture, Transfections and Cell Lines
MDA-MB-231, A549, MCF7, MiaPaca2, HCT 116, U251, HEK 293, HT1080 and 293T cell lines were obtained from ATCC and grown in DMEM (HyClone™, Pittsburgh, PA, USA) supplemented with 10% fetal bovine serum (FBS), 20 mM L-Glutamine and 100 U/mL penicillin/streptomycin. TIME cells were obtained from Dr McMahon and grown in EGM2 medium (Lonza, Basel, Switzerland) supplemented with 2% FBS and 100 U/mL of penicillin/streptomycin. Cells were maintained in a humidified incubator at 37 °C with 5% CO2. ANXA2 depleted cell lines and/or over-expressing H-RasV12 or pBABE were obtained by transfection of Phoenix cells with 4 μg of the plasmids described on table SII, using 8 μL of jetPRIME (Polyplus, Strasbourg, France) transfection reagent according to manufacturers’ instructions. 48 h after transfection the target cells were infected with Phoenix supernatants and selected with 2–5 μg/mL of puromycin for pSUPER-retro-puro or pBABE or 1 mg/mL neomycin for pSUPER-retro-neo backbone plasmids. HT1080 and MDA-MB-231 ANXA2 KO cell lines were obtained by tranfection of the WT cells with 3 µg of pANXA2-gRNA1-px459-V2 or pANXA2-gRNA2-px459-V2 using 6 µL of jetPRIME transfection reagent according to the manufacturers’ instructions. 48 h after transfection the cells were selected with 5 µg/mL of puromycin. Serial dilutions were performed to obtain HT1080 and MDA-MB-231 ANXA2 KO sub-populations. For transient transfection and co-immunoprecipitation experiments, 293T cells in 6 wells plates were transfected for 24–48 h with 1 μg of either pcDNA3, pcDNA3-ANXA2, pcDNA3-ANXA-Cys-8-Ala, pcDNA3-ANXA-Cys-132-Ala, pcDNA3-ANXA-Cys-334-Ala, pcDNA3.1-PTEN, pcDNA3.1-PTEN-Cys-124-Ser, or GFP-PTEN plasmids, described below, using 2 μL (single plasmid transfection) or 4 μL (co-transfection) of jetPRIME transfection reagent according to the manufacturers’ instructions.
4.2. Plasmids and Transfections
Construction of shRNA and CRISPR/Cas9 plasmids are described on
Supplementary Materials Table S2. Construction of pcDNA3-ANXA2, pcDNA3-ANXA2-Cys-8-Ala, pcDNA3-ANXA2-Cys-132-Ala and pcDNA3-ANXA2-Cys-334-Ala plasmids is described in [
1]. PTEN-Wt and PTEN-Cys-124-Ser cDNAs were cloned into pcDNA3.1 vector at restriction sites BamHI/NotI to generate pcDNA3.1-PTEN and pcDNA3.1-PTEN-Cys-124-Ser these plasmids were kindly provided by Dr. Wolfgang Link. GFP-PTEN was a gift from Dr. Alonzo Ross (Addgene plasmid # 13039). The plasmids pBabe-puro (1764) and pBabe-puro H-RasV12 (39526) were purchased from Addgene, Watertown, MA, USA.
ANXA2 depleted cell lines and/or over-expressing H-RasV12 or pBABE were obtained by transfection of Phoenix cells with 4 μg of the plasmids described on
Supplementary Materials Table S2, using 8 μL of jetPRIME (Polyplus) transfection reagent according to manufacturers’ instructions. 48 h after transfection the target cells were infected with Phoenix supernatants and selected with 2–5 μg/mL of puromycin for pSUPER-retro-puro or pBABE or 1 mg/mL neomycin for pSUPER-retro-neo backbone plasmids. HT1080 and MDA-MB-231 ANXA2 KO cell lines were obtained by tranfection of the WT cells with 3 µg of pANXA2-gRNA1-px459-V2 or pANXA2-gRNA2-px459-V2 using 6 µL of jetPRIME transfection reagent according to the manufacturers’ instructions. 48 h after transfection the cells were selected with 5 µg/mL of puromycin. Serial dilutions were performed to obtain HT1080 and MDA-MB-231 ANXA2 KO sub-populations. For transient transfection and co-immunoprecipitation experiments, 293T cells in 6 wells plates were transfected for 24–48 h with 1 μg of either pcDNA3, pcDNA3-ANXA2, pcDNA3-ANXA-Cys-8-Ala, pcDNA3-ANXA-Cys-132-Ala, pcDNA3-ANXA-Cys-334-Ala, pcDNA3.1-PTEN, pcDNA3.1-PTEN-Cys-124-Ser, or GFP-PTEN plasmids, described below, using 2 μL (single plasmid transfection) or 4 μL (co-transfection) of jetPRIME transfection reagent according to the manufacturers’ instructions.
4.4. Immunoprecipitation Assays
For co-immunoprecipitations, cells were washed with PBS, lysed with Triton X-100 lysis buffer (50 mM Tris pH 7.5, 0.2% Triton X-100, 15 mM NaCl, 2 mM EGTA, 1 mM EDTA, protease inhibitors, 5 mM NaVO4, 10 mM NaF) degassed for 1 h. Cell lysates were incubated with specific antibodies for 1 h at 4 °C and then pre-washed 50% slurry protein G-Sepharose™ (GE Healthcare, Pittsburgh, PA, USA) was added for 1 h at 4 °C. Immunoprecipitates were washed four times with 300 μL of lysis buffer, resuspended with 20 μL 2× SDS-PAGE loading buffer, boiled for 10 min, subjected to SDS-PAGE and analyzed by western blotting.
4.5. Western Blotting
Cells were treated as described in the results section and extracts were prepared with lysis buffer (2 mM EGTA, 1 mM EDTA, 50 mM Tris pH 7.5, 120 mM NaCl, 0.1% Na deoxycholate, 1% NP-40, 10 mM NaF, 5 mM NaVO4, protease inhibitors). For western blotting, 20 μg of each cell lysate was subjected to SDS-PAGE, transferred onto a nitrocellulose membrane, incubated with appropriate antibodies and visualized using ChemiDoc™ XRS (BioRad®, Watford, UK).
4.6. qRT-PCR
RNA was isolated using the RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions. The gene expression was measured by qRT-PCR using the One-step NZYSpeedy RT-qPCR Green kit (Nzytech, Lisbon, Portugal) according to manufacturer´s instructions in a Lightcycler 96 instrument (Roche, Basel, Switzerland). Gene expression levels were normalized to RPLP0 mRNA using the 2
−ΔΔCT method [
30]. Error bars represent the standard deviation obtained from average of triplicate samples from three independent experiments. The primers used for qRT-PCR are on
Supplementary Materials Table S4.
4.7. Immunofluorescence Studies
Snap frozen tumors were sectioned on a cryostat and mounted on slides. Slides were fixed for 30 min using −20 °C cold methanol. Slides were incubated in a humidified chamber at 37 °C for 30 min with swine serum (Dako, Glostrup, Denmark). The serum was removed and the slides were incubated with anti-ANXA2 (dil 1:500) and anti-PRDX2 (dil 1:500) for 1 h in a humidified chamber at 37 °C. After washing three times with PBS for 10 min at RT, slides were incubated with the appropriate FITC-conjugated secondary antibodies in a humidified chamber at 37 °C for 1 h. After washing as described above, chamber slides were mounted on glass cover slips using Vector mounting solution for fluorescence (Vector Laboratories, UK) and viewed using a Zeiss LSM 510 META -Laser Scanning Confocal Microscope (Carl Zeiss Inc., Oberkochen, Germany.
4.8. H&E Staining
Snap frozen tumors were sectioned using a cryostat and mounted on slides. Samples were incubated in 95% alcohol for 2 min and 70% alcohol for 2 min. Afterwards they were washed briefly in distilled water. Slides were immersed in Harris hematoxylin solution for 8 min and then washed in running tap water for 5 min. After washing, slides were incubated in 1% acid alcohol for 30 s and washed in running tap water for 1 min. Slides were incubated in 0.2% ammonia water for 45 s and washed again in running tap water for 5 min. Slides were rinsed with 95% alcohol and counterstained in eosin-phloxine B solution 1 min. Following incubation samples were dehydrated with 95% alcohol for 5 min and two changes of absolute alcohol for 5 min. Slides were cleared by 2 changes of xylene for 5 min each. Slides were mounted with xylene based mounting medium (DAKO, Glostrup, Denmark) and visualized using an optical microscope (Axiovert 40C, Carl Zeiss Inc., Oberkochen, Germany).
4.9. Clinical Samples Collection
Colon carcinoma samples were obtained from patients at the time of surgery. Patients´ consent was obtained and the experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report. Ethical approval was obtained for each procedure from the Research Ethics Board, (VG Hospital, Halifax, Nova Scotia, Canada; ethical code: CDHA-RS/2010-111, 17 September 2010).
4.10. Oxidative Stress RT2 PCR Profiler Array
1 × 106 cells were grown in 100 mm plates for 24 h. After what RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, Manchester, UK) according to the manufacturer´s instructions. RNA quality was analyzed using an RNA 6000 Nano Kit chip assay (Agilent, Santa Clara, CA, USA) in a 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) to ensure RNA extracts had an integrity index value greater than 9.0. Two µg of each RNA was used for cDNA synthesis using the RT2 first strand kit (Qiagen, Manchester, UK) according to the manufacturer’s instructions. The RT2 Profiler™ PCR Array Human Oxidative Stress; ref: PAHS-065Z (Qiagen, Manchester, UK) was performed according to the manufacturer’s instructions in a LightCycler 96 instrument (Roche, Basel, Switzerland).
4.11. Statistical Analysis
The statistical significance was evaluated using two-tailed Student’s t-test. The p-values were calculated from at least three independent experiments (n ≥ 3).
4.12. Supplementary Materials and Methods
Materials and Methods related to supplementary Figures are described in
Appendix A.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






