Association Analysis of Deep Genomic Features Extracted by Denoising Autoencoders in Breast Cancer


Abstract
1. Introduction
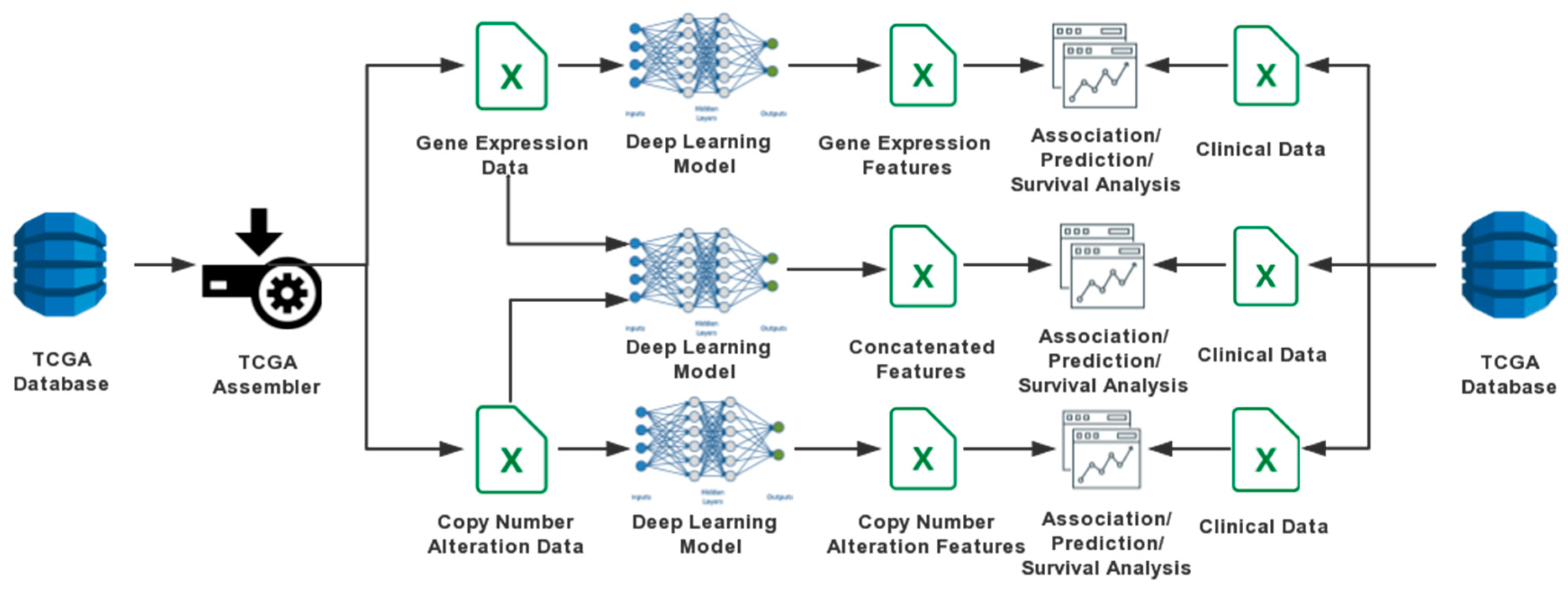
2. Materials and Methods
2.1. Data Sources
2.2. DA Models
2.2.1. One-Input DAs Model
decode = sigmoid (W’ × encode + b’)
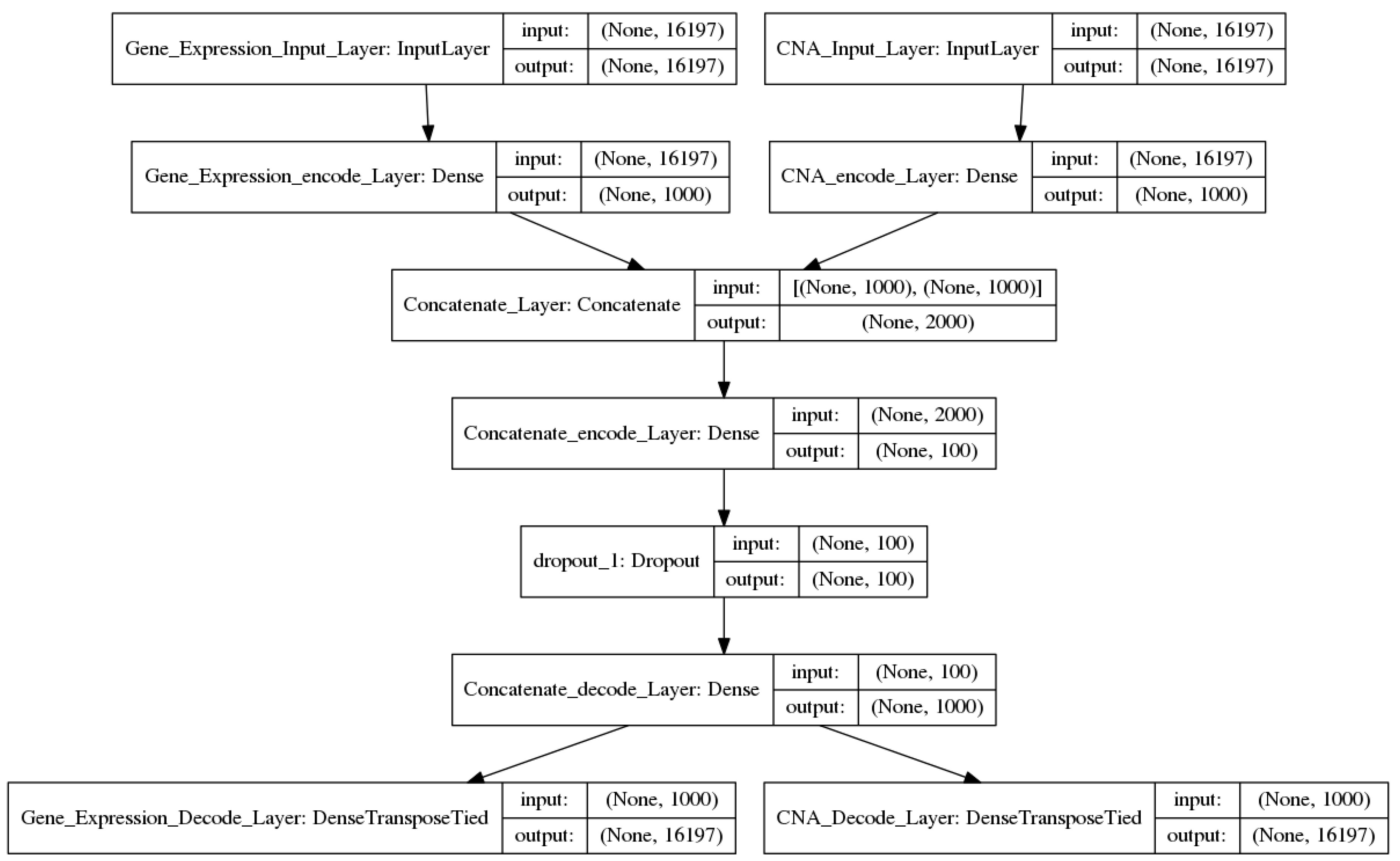
2.2.2. Two-Input DAs Model
input2_encode1 = sigmoid (input2_W1 × input2 + input2_b1)
concate_encode1 = concatenate (input1_encode1, input2_encode1)
concate_encode2 = sigmoid (concate_W2 × concate_encode1+ concate_b2)
output1 = sigmoid (input1_W1′ × concate_encode2 + input1_b1′)
output2 = sigmoid (input2_W1′ × concate_encode2 + input2_b1′)
2.3. Train the Models
2.4. Visualization and Clustering
2.5. Association Analysis
2.6. Gene Sets Enrichment Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DL | Deep learning |
| CNA | Copy number alteration |
| DAs | Denoising autoencoders |
| TCGA | The Cancer Genome Atlas |
| KM | Kaplan-Meier |
| COX-PH | Cox’s proportion hazard |
| OR | Odds ratio |
| AMPK | AMP-activated protein kinase |
| FDR | False discovery rate |
| GSEA | Gene sets enrichment analysis |
| ML | Machine learning |
| ANN | Artificial neural network |
| SVM | Support vector machine |
| ER | Estrogen receptor |
| miRNA | Micro RNA |
| CPM | Count per million |
| FPKM-UQ | Upper quartile fragments per kilobase of transcript per Million mapped reads |
| SGD | Stochastic gradient descent |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
References
- Lesk, A.M. Introduction to Bioinformatics, 3rd ed.; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Bergamaschi, A.; Kim, Y.H.; Wang, P.; Sørlie, T.; Hernandez-Boussard, T.; Lonning, P.E.; Tibshirani, R.; Borresen-Dale, A.L.; Pollack, J.R. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer 2006, 45, 1033–1040. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Tamayo, P.; Rifkin, R.; Mukherjee, S.; Yeang, C.H.; Angelo, M.; Ladd, C.; Reich, M.; Latulippe, E.; Mesirov, J.P.; et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. USA 2001, 98, 15149–15154. [Google Scholar] [CrossRef]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.T.; Hajirasouliha, I.; Raphael, B.J. Detecting independent and recurrent copy number aberrations using interval graphs. Bioinformatics 2014, 30, i195–i203. [Google Scholar] [CrossRef][Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Boughorbel, S.; Al-Ali, R.; Elkum, N. Model Comparison for Breast Cancer Prognosis Based on Clinical Data. PLoS ONE 2016, 11, e0146413. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Parker, J.S.; Leung, S.; Voduc, D.; Ebbert, M.; Vickery, T.; Davies, S.R.; Snider, J.; Stijleman, I.J.; Reed, J.; et al. A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen-treated estrogen receptor-positive breast cancer. Clin. Cancer Res. 2010, 16, 5222–5232. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Murphy, L.C.; Hu, P. Recurrent copy number alterations in young women with breast cancer. Oncotarget 2018, 9, 11541–11558. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Auria, L.; Moro, R.A. Support Vector Machines (SVM) as a Technique for Solvency Analysis; DIW Discussion Papers 811; DIW Berlin, German Institute for Economic Research: Berlin, Germany, 2008. [Google Scholar]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Schadt, E.E.; Lamb, J.; Yang, X.; Zhu, J.; Edwards, S.; Guhathakurta, D.; Sieberts, S.K.; Monks, S.; Reitman, M.; Zhang, C.; et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat. Genet. 2005, 37, 710–717. [Google Scholar] [CrossRef]
- Tan, J.; Ung, M.; Cheng, C.; Greene, C.S. Unsupervised feature construction and knowledge extraction from genome-wide assays of breast cancer with denoising autoencoders. Pac. Symp. Biocomput. 2014, 20, 132–143. [Google Scholar]
- Guyon, I.; Elisseeff, A. Feature Extraction, Foundations and Applications: An introduction to feature extraction. Stud. Fuzziness Soft Comput. 2006, 207, 1–25. [Google Scholar]
- Vincent, P.; Larochelle, H.; Bengio, Y.; Manzagol, P.A. Extracting and composing robust features with denoising autoencoders. In Proceedings of the 25th International Conference on Machine Learning, Helsinki, Finland, 5–9 July 2008; ACM: New York, NY, USA, 2008; pp. 1096–1103. [Google Scholar]
- Khan, J.; Wei, J.S.; Ringnér, M.; Saal, L.H.; Ladanyi, M.; Westermann, F.; Berthold, F.; Schwab, M.; Antonescu, C.R.; Peterson, C.; et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat. Med. 2001, 7, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Oliver, S. Deep Learning for Computational Biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Wspolczesna Onkol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qiu, P.; Ji, Y. TCGA-assembler: Open-source software for retrieving and processing TCGA data. Nat. Methods 2014, 11, 599–600. [Google Scholar] [CrossRef]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Chollet, F. Building Autoencoders in Keras. The Keras Blog. 2016. Available online: https://blog.keras.io/building-autoencoders-in-keras.html (accessed on 20 January 2019).
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A System for Large-Scale Machine Learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI’16), Savannah, GA, USA, 2–4 November 2016; pp. 265–284. [Google Scholar]
- Wu, G.; Xing, M.; Mambo, E.; Huang, X.; Liu, J.; Guo, Z.; Chatterjee, A.; Goldenberg, D.; Gollin, S.M.; Sukumar, S.; et al. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005, 7, R609–R616. [Google Scholar] [CrossRef]
- Ching, T.; Zhu, X.; Garmire, L.X. Cox—Nnet: An artificial neural network method for prognosis prediction on high—Throughput omics data. PLoS Comput. Biol. 2016, 14, e1006076. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Giordanetto, F.; Karis, D. Direct AMP-activated protein kinase activators: A review of evidence from the patent literature. Expert Opin. Ther. Pat. 2012, 22, 1467–1477. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Harris, L.; Fritsche, H.; Mennel, R.; Norton, L.; Ravdin, P.; Taube, S.; Somerfield, M.R.; Hayes, D.F.; Bast, R.C., Jr. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J. Clin. Oncol. 2007, 25, 5287–5312. [Google Scholar] [CrossRef]
- Miyahara, E.; Toi, M.; Wada, T.; Yamada, H.; Osaki, A.; Yanagawa, E.; Toge, T. The expression of NCC-ST-439, a tumor marker, in human breast cancer patients. Gan No Rinsho 1990, 36, 2023–2026. [Google Scholar]
- Mobadersany, P.; Yousefi, S.; Amgad, M.; Gutman, D.A.; Barnholtz-Sloan, J.S.; Vega, J.E.V.; Brat, D.J.; Cooper, L.A.D. Predicting cancer outcomes from histology and genomics using convolutional networks. Proc. Natl. Acad. Sci. USA 2018, 115, E2970–E2979. [Google Scholar] [CrossRef]
- Wang, E.; Zaman, N.; Mcgee, S.; Milanese, J.S.; Masoudi-Nejad, A.; O’Connor-McCourt, M. Predictive genomics: A cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin. Cancer Biol. 2015, 30, 4–12. [Google Scholar] [CrossRef]
- Vincent, P.; Larochelle, H.; Lajoie, I.; Bengio, Y.; Manzagol, P.A. Stacked denoising autoencoders: Learning useful representations in a deep network with a local denoising criterion. J. Mach. Learn. Res. 2010, 11, 3371–3408. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Data Source | Deep Features (Noise Factors = 0.25) | |
|---|---|---|---|
| One-input DAs | Gene expressions | Activity values | 1085 × 100 |
| weights | 16,197 × 100 | ||
| Copy number alterations | Activity values | 1085 × 100 | |
| weights | 16,197 × 100 | ||
| Two-input DAs | Gene expressions Copy number alterations | Activity values | 1085 × 100 |
| weights | 16,197 × 100 | ||
| Clinical Characteristics | Fisher’s Exact p-Value | Chi-Square Test p-Value |
|---|---|---|
| Pathological T | 0.69 | 0.69 |
| Pathological N | 0.95 | 0.96 |
| Pathological M | 0.95 | 0.94 |
| Tumor Stage | 0.93 | 0.93 |
| ER Status | 0.002 | 0.002 |
| PR Status | 1.00 | 0.99 |
| HER Status | 0.43 | 0.44 |
| Age * | 0.58 | 0.67 |
| Triple Negative Status | 0.15 | 0.17 |
| Tumor Subtype | 0.35 | 0.36 |
| Risk Score | HR | Lower.95_HR | Upper.95_HR | p-Value |
|---|---|---|---|---|
| Gene expression | 1.009 | 1.005 | 1.013 | 1.06 × 10−5 |
| CNA | 1.23 | 1.15 | 1.32 | 7.86 × 10−9 |
| Concatenated | 1.27 | 1.16 | 1.40 | 5.62 × 10−7 |
| Gene Sets | p-Value | Adjusted p-Value |
|---|---|---|
| regulation of transcription, DNA-templated | 2.25 × 10−7 | 0.001 |
| regulation of nucleic acid-templated transcription | 6.03 × 10−5 | 0.04 |
| regulation of apoptotic process | 6.19 × 10−5 | 0.04 |
| positive regulation of gene expression | 5.89 × 10−5 | 0.04 |
| positive regulation of cell proliferation | 4.78 × 10−5 | 0.04 |
| AMPK signaling pathway_Homo sapiens_hsa04152 | 6.08 × 10−5 | 0.018 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Hu, P. Association Analysis of Deep Genomic Features Extracted by Denoising Autoencoders in Breast Cancer. Cancers 2019, 11, 494. https://doi.org/10.3390/cancers11040494
Liu Q, Hu P. Association Analysis of Deep Genomic Features Extracted by Denoising Autoencoders in Breast Cancer. Cancers. 2019; 11(4):494. https://doi.org/10.3390/cancers11040494
Chicago/Turabian StyleLiu, Qian, and Pingzhao Hu. 2019. "Association Analysis of Deep Genomic Features Extracted by Denoising Autoencoders in Breast Cancer" Cancers 11, no. 4: 494. https://doi.org/10.3390/cancers11040494
APA StyleLiu, Q., & Hu, P. (2019). Association Analysis of Deep Genomic Features Extracted by Denoising Autoencoders in Breast Cancer. Cancers, 11(4), 494. https://doi.org/10.3390/cancers11040494