HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action

 ,
,
Abstract
1. Introduction
2. HDAC Inhibitors in the Clinic
3. Roles of Each HDAC in the Treatment of MM (Expression, Prognostic Significance, Molecular Mechanisms and Therapeutic Targets Revealed by Preclinical Studies)
3.1. HDAC1 and HDAC3 (Class I)
3.2. HDAC4 (Class IIa)
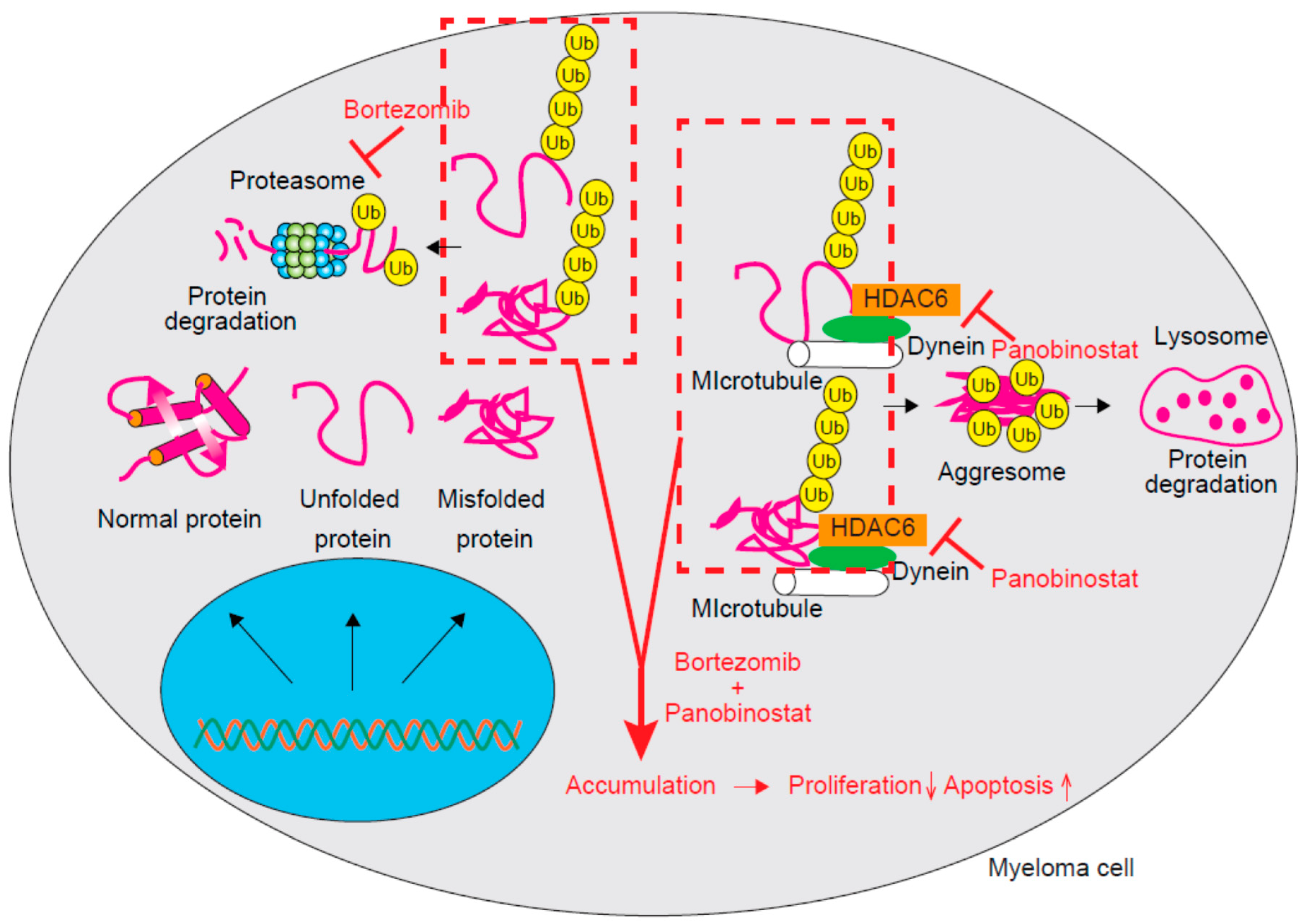
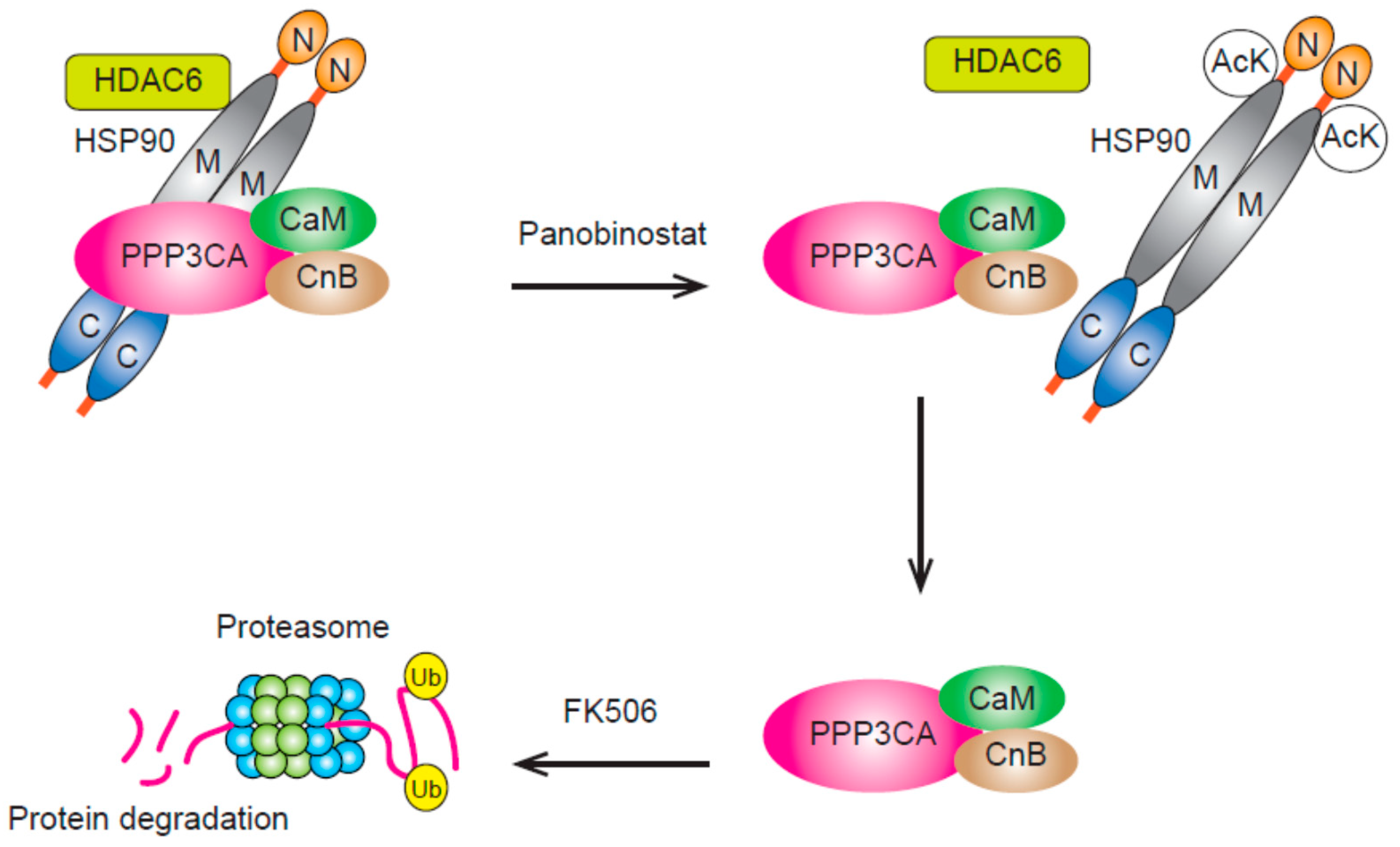
3.3. HDAC6 and HDAC10 (Class IIb)
3.4. HDAC11 (Class IV)
4. Strategy for Enhancing the Anti-Myeloma Effect of HDAC Inhibitors by Adding Other Agents
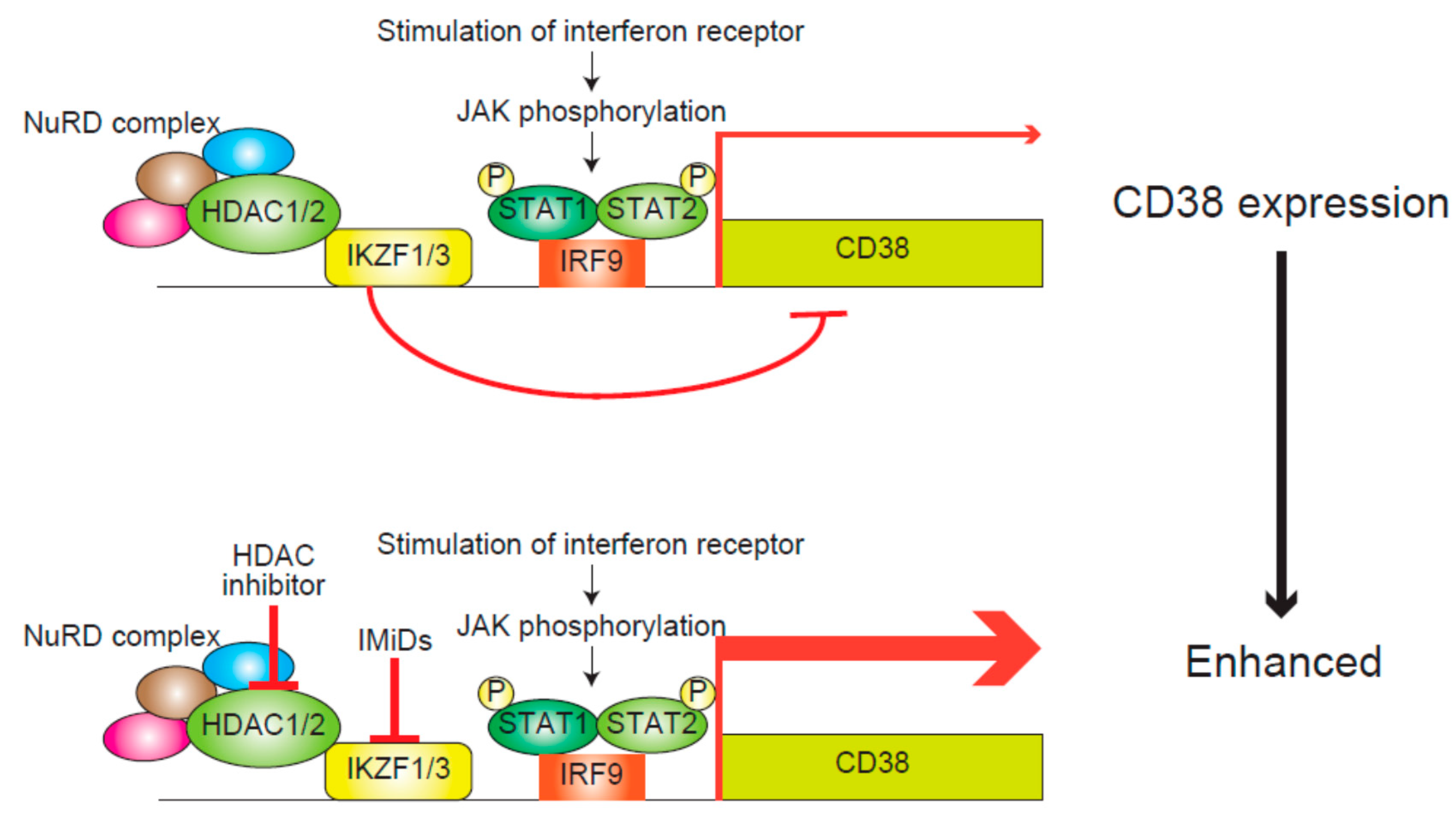
4.1. IMiDs
4.2. Cladribine (2CdA)
4.3. PI3K Inhibitor
4.4. Dual Inhibition of HDAC and BCL-XL
4.5. mTOR Inhibitors
4.6. 5-Azacytidine (AZA)
5. Modification of Immune Functions by HDAC Inhibitors for the Treatment of Myeloma
6. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HDACs | histone deacetylases |
| MM | multiple myeloma |
| PPP3CA | protein phosphatase 3 catalytic subunit α |
| HSP90 | heat shock protein 90 |
| IMiDs | immunomodulatory drugs |
| RUNX2 | runt-related transcription factor 2 |
| CBFA1 | core binding factor α1 |
| GFI1 | growth factor independent 1 transcriptional repressor |
| EZH2 | enhancer of zeste homolog 2 |
| MST1 | macrophage-stimulating 1 |
| SAPK | stress-activated protein kinase |
| JNK | Janus kinase |
| RASSF | ras association domain family member |
| ESR1 | estrogen receptor 1 |
| CDK6 | Cell division protein kinase 6 |
| IRF4 | interferon regulatory factor 4 |
| NFATc1 | nuclear factor of activated T-cells, cytoplasmic 1 |
| NF-κB | nuclear factor-kappa B |
| 17-AAG | 17-N-allylamino-17-demethoxygeldanamycin |
| APCs | antigen-presenting cells |
| IL-10 | interleukin-10 |
| MDSCs | myeloid-derived suppressor cells |
| WT | wild-type |
| CRBN | cereblon |
| CRL4 | Cullin-RING ubiquitin ligase 4 |
| IKZF1/3 | Ikaros family zinc finger protein 1/3 |
| 2CdA | cladribine |
| Chk2 | checkpoint kinase 2 |
| PI3K | phosphoinositide 3-kinase |
| IL-6 | interleukin-6 |
| IGF-1 | insulin-like growth factor-1 |
| RARRES3 | retinoic acid receptor responder 3 |
| B-CLL | B cell chronic lymphocytic leukemia |
| BCL-XL | B-cell lymphoma-extra large |
| ATF-3 | activating transcription factor 3 |
| AZA | 5-azacytidine |
| DNMT | DNA methyltransferases |
| PD-L1 | programmed cell death ligand 1 |
| PD-1 | programmed cell death 1 |
| STAT3 | signal transducers and activators of transcription |
| MHC | major histocompatibility complex |
| NK | natural killer |
| MICA | major histocompatibility complex class I-related chain A |
| MICB | major histocompatibility complex class I-related chain B |
References
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Tashima, T.; Murata, H.; Kodama, H. Design and synthesis of novel and highly-active pan-histone deacetylase (pan-HDAC) inhibitors. Bioorg. Med. Chem. 2014, 22, 3720–3731. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Kato, N.; Tanaka, J.; Sugita, J.; Toubai, T.; Miura, Y.; Ibata, M.; Syono, Y.; Ota, S.; Kondo, T.; Asaka, M.; et al. Regulation of the expression of MHC class I-related chain A, B (MICA, MICB) via chromatin remodeling and its impact on the susceptibility of leukemic cells to the cytotoxicity of NKG2D-expressing cells. Leukemia 2007, 21, 2103–2108. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Richardson, P.G.; Schlossman, R.L.; Alsina, M.; Weber, D.M.; Coutre, S.E.; Gasparetto, C.; Mukhopadhyay, S.; Ondovik, M.S.; Khan, M.; Paley, C.S.; et al. PANORAMA 2: Panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013, 122, 2331–2337. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kronenberg, H. Minireview: Transcriptional regulation in development of bone. Endocrinology 2005, 146, 1012–1017. [Google Scholar] [CrossRef]
- D’Souza, S.; del Prete, D.; Jin, S.; Sun, Q.; Huston, A.J.; Kostov, F.E.; Sammut, B.; Hong, C.S.; Anderson, J.L.; Patrene, K.D.; et al. Gfi1 expressed in bone marrow stromal cells is a novel osteoblast suppressor in patients with multiple myeloma bone disease. Blood 2011, 118, 6871–6880. [Google Scholar] [CrossRef]
- Adamik, J.; Jin, S.; Sun, Q.; Zhang, P.; Weiss, K.R.; Anderson, J.L.; Silbermann, R.; Roodman, G.D.; Galson, D.L. EZH2 or HDAC1 Inhibition Reverses Multiple Myeloma-Induced Epigenetic Suppression of Osteoblast Differentiation. Mol. Cancer Res. 2017, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Thiagalingam, S.; Cheng, K.H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone deacetylases: Unique players in shaping the epigenetic histone code. Ann. N. Y. Acad. Sci. 2003, 983, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Richardson, P.G.; McMullan, C.; Poulaki, V.; Fanourakis, G.; Schlossman, R.; Chauhan, D.; Munshi, N.C.; Hideshima, T.; et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood 2003, 101, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.Y.; Dai, Y.; Grant, S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.Y.; Ngo, L.; Xu, W.S.; Richon, V.M.; Marks, P.A. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc. Natl. Acad. Sci. USA 2004, 101, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, H.; Khayat, D.; Giaccone, G.; Facon, T. Proteasome inhibition and its clinical prospects in the treatment of hematologic and solid malignancies. Cancer 2005, 104, 1794–1807. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, E.; Maes, K.; Verhulst, S.; Lui, H.; Kassambara, A.; Maes, A.; Robert, N.; Heirman, C.; Cakana, A.; Hose, D.; et al. Loss of RASSF4 Expression in Multiple Myeloma Promotes RAS-Driven Malignant Progression. Cancer Res. 2018, 78, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Harding, T.; Swanson, J.; Van Ness, B. EZH2 inhibitors sensitize myeloma cell lines to panobinostat resulting in unique combinatorial transcriptomic changes. Oncotarget 2018, 9, 21930–21942. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Stamato, M.A.; Gullà, A.M.; Morelli, E.; Romeo, E.; Raimondi, L.; Pitari, M.R.; Ferrandino, I.; Misso, G.; Caraglia, M.; et al. Therapeutic Targeting of miR-29b/HDAC4 Epigenetic Loop in Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 1364–1375. [Google Scholar] [CrossRef]
- Stamato, M.A.; Juli, G.; Romeo, E.; Ronchetti, D.; Arbitrio, M.; Caracciolo, D.; Neri, A.; Tagliaferri, P.; Tassone, P.; Amodio, N. Inhibition of EZH2 triggers the tumor suppressive miR-29b network in multiple myeloma. Oncotarget 2017, 8, 106527. [Google Scholar] [CrossRef]
- Plemper, R.K.; Wolf, D.H. Retrograde protein translocation: ERADication of secretory proteins in health and disease. Trends Biochem. Sci. 1999, 24, 266–270. [Google Scholar] [CrossRef]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- LeBlanc, R.; Catley, L.P.; Hideshima, T.; Lentzsch, S.; Mitsiades, C.S.; Mitsiades, N.; Neuberg, D.; Goloubeva, O.; Pien, C.S.; Adams, J.; et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002, 62, 4996–5000. [Google Scholar] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef]
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167. [Google Scholar] [CrossRef]
- Bat-Erdene, A.; Miki, H.; Oda, A.; Nakamura, S.; Teramachi, J.; Amachi, R.; Tenshin, H.; Hiasa, M.; Iwasa, M.; Harada, T.; et al. Synergistic targeting of Sp1, a critical transcription factor for myeloma cell growth and survival, by panobinostat and proteasome inhibitors. Oncotarget 2016, 7, 79064–79075. [Google Scholar] [CrossRef]
- Imai, Y.; Ohta, E.; Takeda, S.; Sunamura, S.; Ishibashi, M.; Tamura, H.; Wang, Y.H.; Deguchi, A.; Tanaka, J.; Maru, Y.; et al. Histone deacetylase inhibitor panobinostat induces calcineurin degradation in multiple myeloma. JCI Insight 2016, 1, e85061. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Deb, J.; Patra, A.K.; Thuy Pham, D.A.; Chen, W.; Vaeth, M.; Berberich-Siebelt, F.; Klein-Hessling, S.; Lamperti, E.D.; Reifenberg, K.; et al. NFATc1 affects mouse splenic B cell function by controlling the calcineurin--NFAT signaling network. J. Exp. Med. 2011, 208, 823–839. [Google Scholar] [CrossRef]
- Medyouf, H.; Alcalde, H.; Berthier, C.; Guillemin, M.C.; dos Santos, N.R.; Janin, A.; Decaudin, D.; de Thé, H.; Ghysdael, J. Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat. Med. 2007, 13, 736–741. [Google Scholar] [CrossRef]
- Cheng, J.; Tang, W.; Su, Z.; Wei, Q. Mutation of calcineurin subunit B M118 influences the activities of NF-AT and p53, but not calcineurin expression level. Biochem. Biophys. Res. Commun. 2011, 413, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Mitsiades, C.S.; Laubach, J.P.; Lonial, S.; Chanan-Khan, A.A.; Anderson, K.C. Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br. J. Haematol. 2011, 152, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Oehme, I.; Linke, J.P.; Böck, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Sun, L.; Aramsangtienchai, P.; Spiegelman, N.A.; Zhang, X.; Huang, W.; Seto, E.; Lin, H. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed]
- Villagra, A.; Cheng, F.; Wang, H.W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [Google Scholar] [CrossRef]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef]
- Sahakian, E.; Powers, J.J.; Chen, J.; Deng, S.L.; Cheng, F.; Distler, A.; Woods, D.M.; Rock-Klotz, J.; Sodre, A.L.; Youn, J.I.; et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol. Immunol. 2015, 63, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Cottini, F.; Ohguchi, H.; Jakubikova, J.; Gorgun, G.; Mimura, N.; Tai, Y.T.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Rational combination treatment with histone deacetylase inhibitors and immunomodulatory drugs in multiple myeloma. Blood Cancer J. 2015, 5, e312. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Fink, E.C.; Ebert, B.L. The novel mechanism of lenalidomide activity. Blood 2015, 126, 2366–2369. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Ma, D.; Cheng, B.; Fang, Q.; Kuang, X.; Yu, K.; Wang, W.; Hu, B.; Wang, J. Crucial role of HO-1/IRF4-dependent apoptosis induced by panobinostat and lenalidomide in multiple myeloma. Exp. Cell Res. 2018, 363, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Lyu, H.; Pei, S.; Song, D.; Ni, J.; Liu, B. Cladribine in combination with entinostat synergistically elicits anti-proliferative/anti-survival effects on multiple myeloma cells. Cell Cycle 2018, 17, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, N.; Ting Lee, J.X.; Adina Nee, H.F.; Bi, C.; Chung, T.H.; Hart, S.; Chng, W.J. VS-5584 mediates potent anti-myeloma activity via the upregulation of a class II tumor suppressor gene, RARRES3 and the activation of Bim. Oncotarget 2017, 8, 101847–101864. [Google Scholar] [CrossRef] [PubMed]
- Casanova, B.; de la Fuente, M.T.; Garcia-Gila, M.; Sanz, L.; Silva, A.; Garcia-Marco, J.A.; Garcia-Pardo, A. The class II tumor-suppressor gene RARRES3 is expressed in B cell lymphocytic leukemias and down-regulated with disease progression. Leukemia 2001, 15, 1521–1526. [Google Scholar] [CrossRef] [PubMed]
- Chüeh, A.C.; Tse, J.W.T.; Dickinson, M.; Ioannidis, P.; Jenkins, L.; Togel, L.; Tan, B.; Luk, I.; Davalos-Salas, M.; Nightingale, R.; et al. ATF3 Repression of BCL-X. Clin. Cancer Res. 2017, 23, 5573–5584. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.K.; Michalowski, A.M.; Gamache, B.J.; DuBois, W.; Patel, J.; Zhang, K.; Gary, J.; Zhang, S.; Gaikwad, S.; Connors, D.; et al. Cooperative Targets of Combined mTOR/HDAC Inhibition Promote MYC Degradation. Mol. Cancer Ther. 2017, 16, 2008–2021. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Ohguchi, H.; Grondin, Y.; Kikuchi, S.; Sagawa, M.; Tai, Y.T.; Mazitschek, R.; Hideshima, T.; Anderson, K.C. HDAC3 regulates DNMT1 expression in multiple myeloma: Therapeutic implications. Leukemia 2017, 31, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Tan, X.F.; Zhang, S.; Wu, T.; Zhang, Z.M.; Ai, H.W.; Song, J. An Intramolecular Interaction of UHRF1 Reveals Dual Control for Its Histone Association. Structure 2018, 26, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Woan, K.V.; Lienlaf, M.; Perez-Villaroel, P.; Lee, C.; Cheng, F.; Knox, T.; Woods, D.M.; Barrios, K.; Powers, J.; Sahakian, E.; et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: Enhanced antitumor immunity and impaired cell proliferation. Mol. Oncol. 2015, 9, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Hideshima, T.; Tai, Y.T.; Song, Y.; Richardson, P.; Raje, N.; Munshi, N.C.; Anderson, K.C. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia 2018, 32, 1932–1947. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Das, D.S.; Song, Y.; Hideshima, T.; Tai, Y.T.; Chauhan, D.; Anderson, K.C. Combination of a novel HDAC6 inhibitor ACY-241 and anti-PD-L1 antibody enhances anti-tumor immunity and cytotoxicity in multiple myeloma. Leukemia 2018, 32, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Nwangwu, C.A.; Weiher, H.; Schmidt-Wolf, I.G.H. Increase of CIK cell efficacy by upregulating cell surface MICA and inhibition of NKG2D ligand shedding in multiple myeloma. Hematol. Oncol. 2017, 35, 719–725. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef]
- Fedele, P.L.; Willis, S.N.; Liao, Y.; Low, M.S.; Rautela, J.; Segal, D.H.; Gong, J.N.; Huntington, N.D.; Shi, W.; Huang, D.C.S.; et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood 2018, 132, 2166–2178. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| HDAC | Class | Substrate | Biological Function | IC50 | ||
|---|---|---|---|---|---|---|
| Panobinostat | Ricolinostat | Vorinostat | ||||
| HDAC1 | I | Histone, p53, GATA1/2, STAT1/3, C/EBPα | Epigenetic regulation Alteration of activity of transcription factors Dysregulation of signaling pathways | ≤10 nM | ≤100 nM and >10 nM | ≤100 nM and >10 nM |
| HDAC2 | I | Histone, GATA1/2, STAT1/3, C/EBPα | Epigenetic regulation Alteration of activity of transcription factors Dysregulation of signaling pathways | ≤100 nM and >10 nM | ≤100 nM and >10 nM | ≤100 nM and >10 nM |
| HDAC3 | I | Histone, GATA1/2, STAT1/3, C/EBPα | Epigenetic regulation Alteration of activity of transcription factors Dysregulation of signaling pathways | ≤10 nM | ≤100 nM and >10 nM | ≤1000 nM and >100 nM |
| HDAC4 | IIa | GATA1/2, STAT1/3, C/EBPα | Epigenetic regulation Alteration of activity of transcription factors Dysregulation of signaling pathways | ≤1000 nM and >100 nM | >1000 nM | ≤1000 nM and >100 nM |
| HDAC6 | IIb | Tubulin, HSP90 | Inhibition of protein degradation Dysregulation of signaling pathways | ≤100 nM and >10 nM | ≤10 nM | ≤1000 nM and >100 nM |
| HDAC8 | I | SMC3 | Epigenetic regulation | ≤1000 nM and >100 nM | ≤100 nM and >10nM | ≤1000 nM and >100 nM |
| HDAC9 | IIa | GATA1/2, STAT1/3, C/EBPα | Epigenetic regulation Alteration of activity of transcription factors Dysregulation of signaling pathways | ≤10 nM | >1000 nM | ≤100 nM and >10 nM |
| Agent | HDAC Inhibitor to Be Combined with Each Agent | Target Molecule | Reference |
|---|---|---|---|
| IMiDs | entinostat, ricolinostat, panobinostat | c-Myc, HO-1, IRF4, c-Myc | Hideshima et al. 2015 [42], Tang et al. 2018 [45] |
| Cladribine | entinostat | cyclin D1, E2F-1, p21waf−1 | Wang et al. 2018 [46] |
| PI3K inhibitor | panobinostat | RARRES3 | Mustafa et al. 2017 [47] |
| mTOR inhibitors | entinostat | Myc, E2F | Simmons et al. 2017 [50] |
| 5-azacytidine | BG45 (HDAC3-selective inhibitor) | DNMT1 | Harada et al. 2017 [51] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imai, Y.; Hirano, M.; Kobayashi, M.; Futami, M.; Tojo, A. HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action. Cancers 2019, 11, 475. https://doi.org/10.3390/cancers11040475
Imai Y, Hirano M, Kobayashi M, Futami M, Tojo A. HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action. Cancers. 2019; 11(4):475. https://doi.org/10.3390/cancers11040475
Chicago/Turabian StyleImai, Yoichi, Mitsuhito Hirano, Masayuki Kobayashi, Muneyoshi Futami, and Arinobu Tojo. 2019. "HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action" Cancers 11, no. 4: 475. https://doi.org/10.3390/cancers11040475
APA StyleImai, Y., Hirano, M., Kobayashi, M., Futami, M., & Tojo, A. (2019). HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action. Cancers, 11(4), 475. https://doi.org/10.3390/cancers11040475