Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/p53 Mutant Lung Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
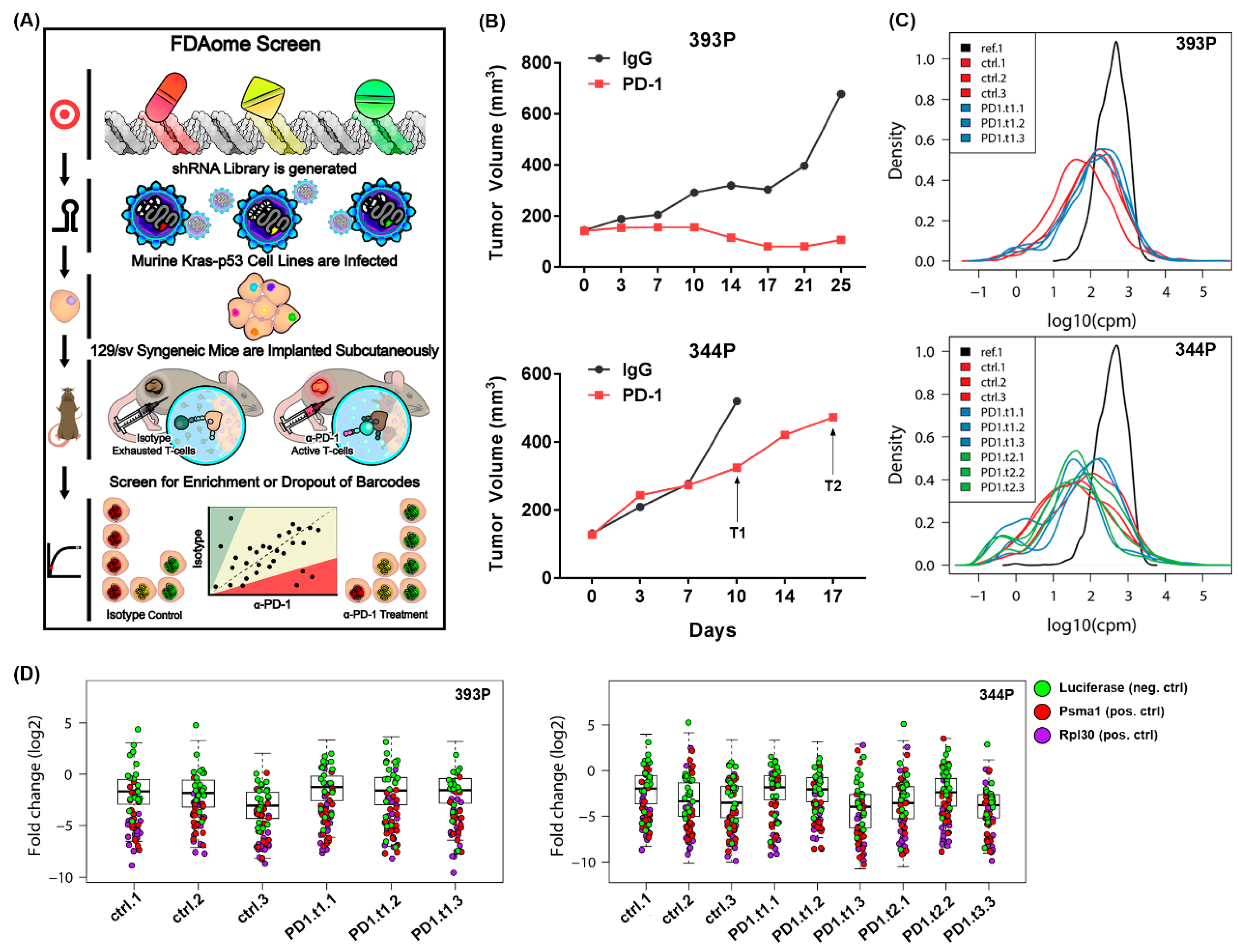
2.1. An In Vivo Functional Genomics Screen to Identify Novel Tumor Cell Vulnerabilities in the Face of Immune Checkpoint Blockade
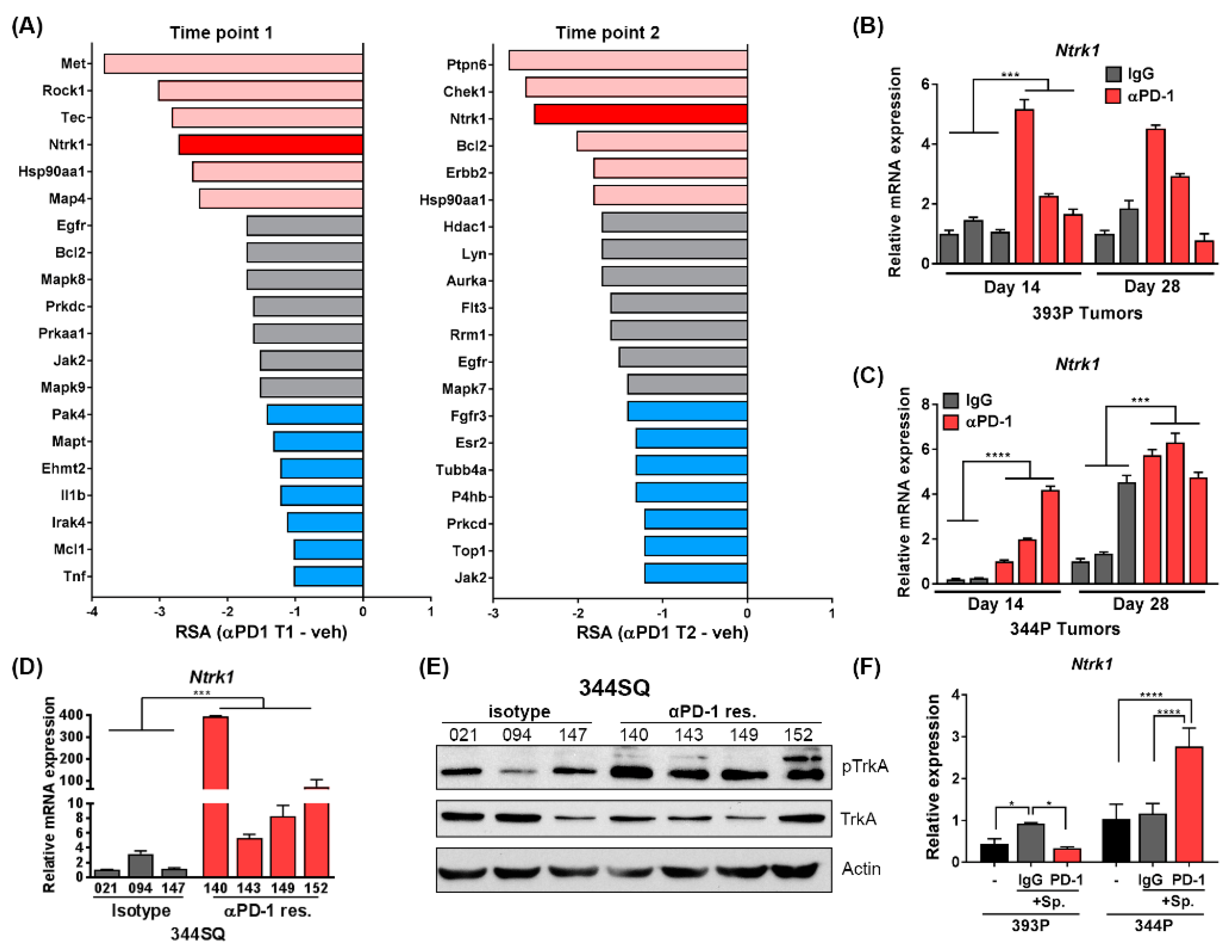
2.2. Short Hairpin RNAs Targeting Ntrk1 Dropped out Significantly from 344P Mesenchymal Tumors Treated with PD-1 Blocking Antibody
2.3. Transcript Level and Protein Activity of Ntrk1 Are Increased in PD-1 Treated Tumors
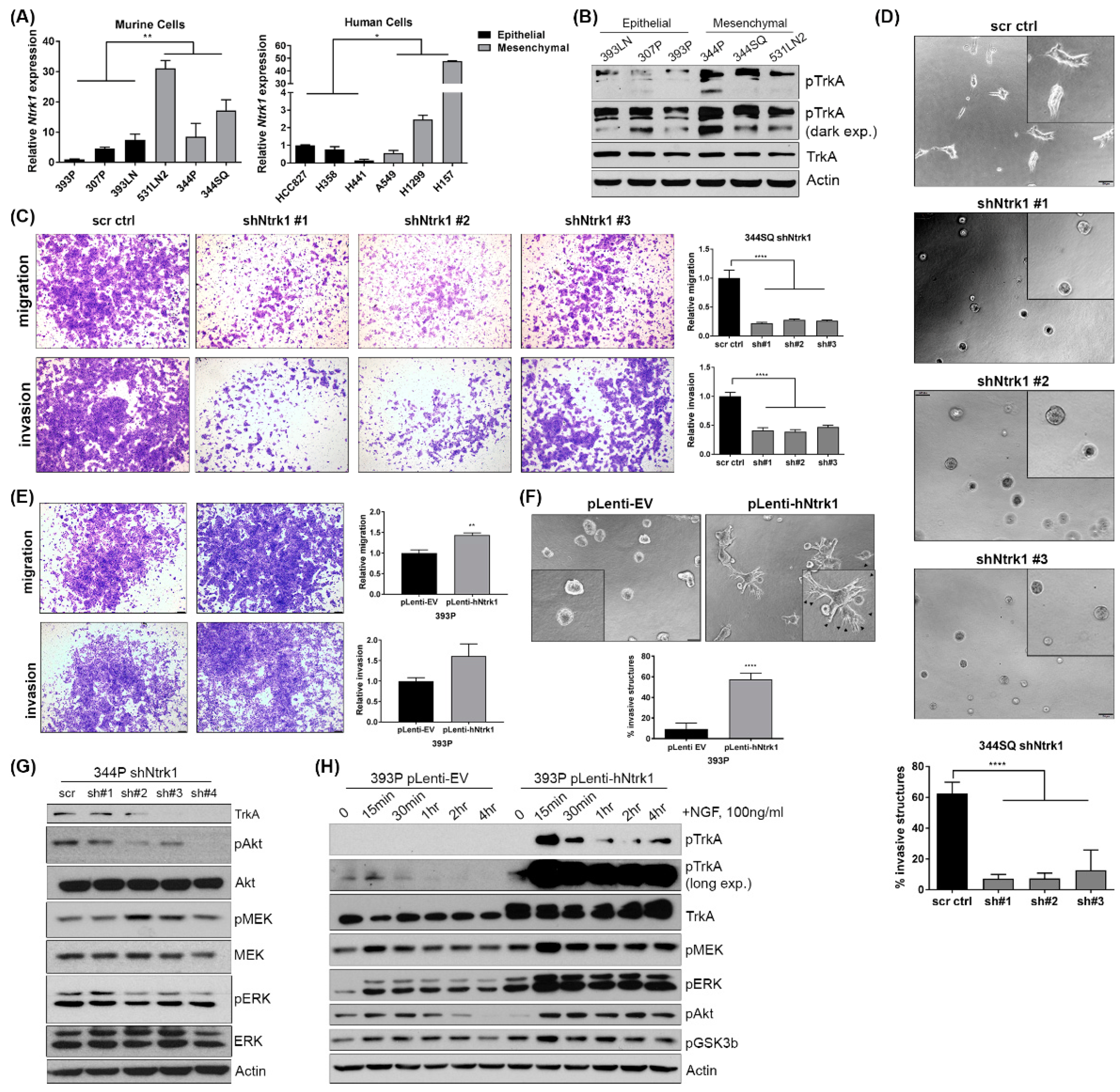
2.4. Baseline Expression of Ntrk1 Is Higher in Mesenchymal Murine and Human Lung Cancer Cell Lines and Is Necessary for Invasion and Migration
2.5. Ntrk1 Activates AKT and MAPK Signaling in KP Lung Cancer Cell Lines to Regulate Cell Growth
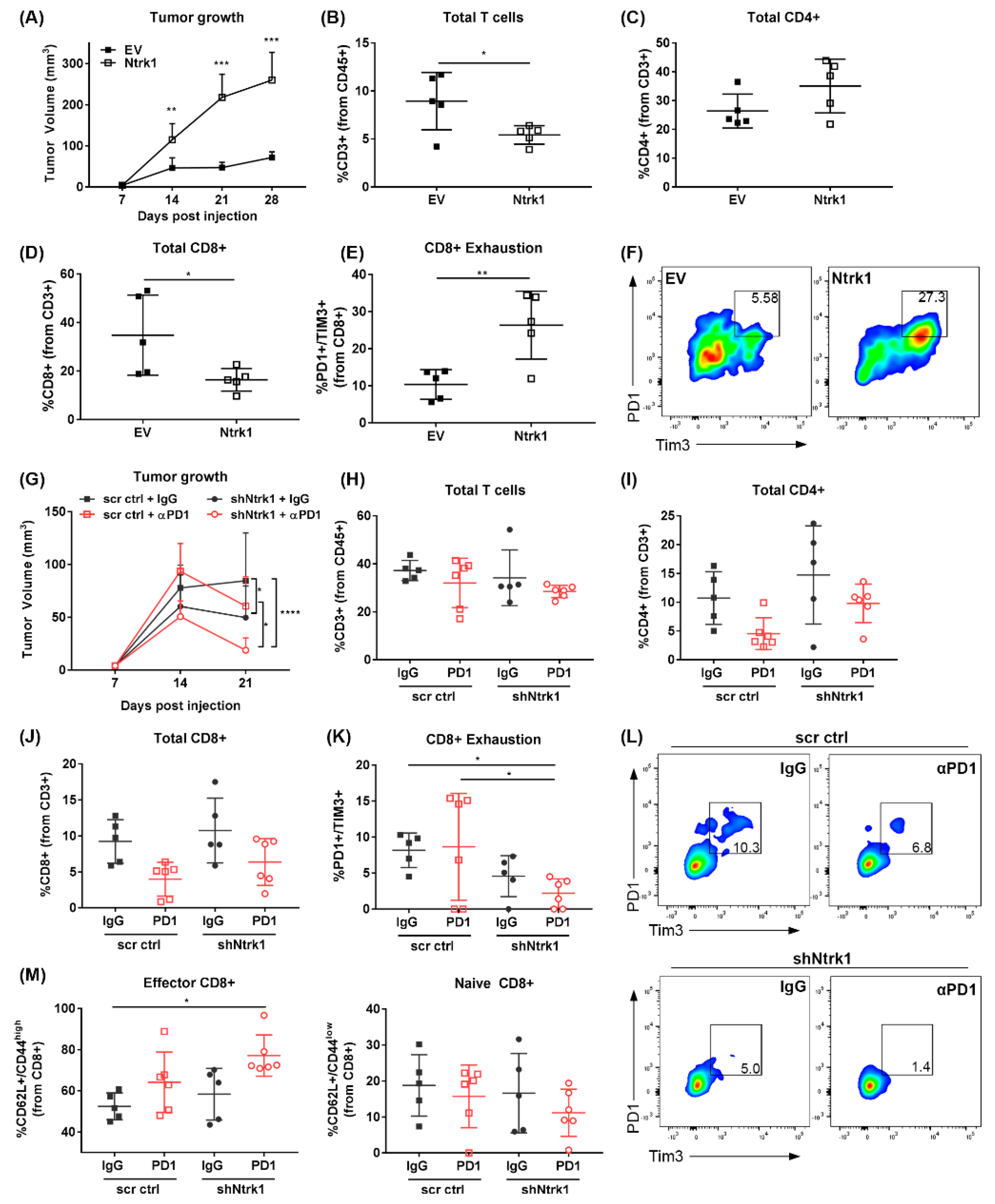
2.6. Ntrk1 Overexpression Promotes Tumor Growth In Vivo and Augments the Tumor Infiltrating Immune Compartment by Promoting CD8+ T Cell Exhaustion
2.7. Combination of Ntrk1 Knockdown with PD-1 Blockade Significantly Reduces Tumor Growth and CD8+ T Cell Exhaustion In Vivo
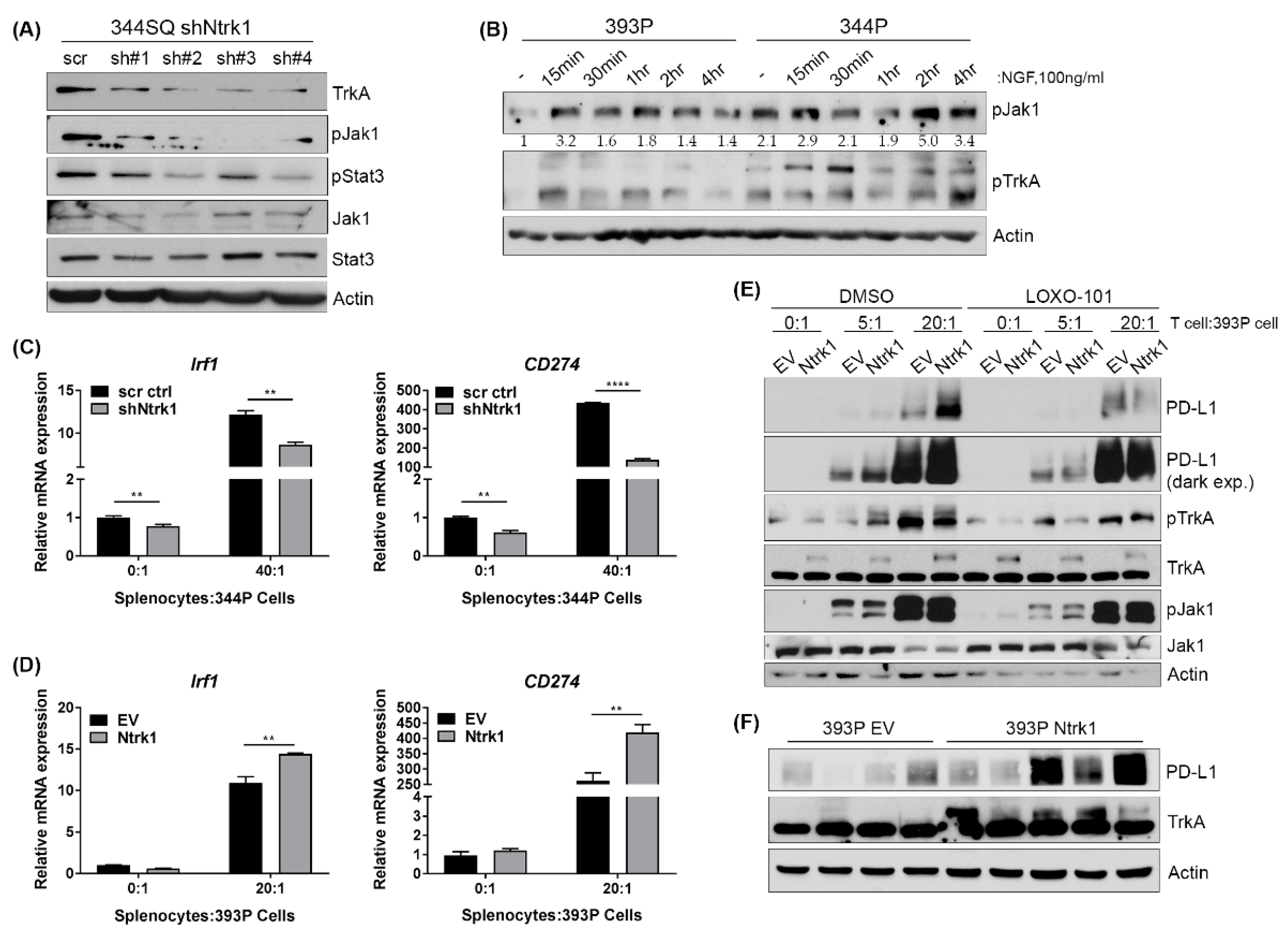
2.8. Ntrk1 Expression Impacts Jak Signaling and Correlates with PD-L1 Levels in Cells Co-Cultured with an Immune Compartment and in Tumor Samples
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Animal Studies
4.3. FDAome Dropout Screen
4.4. Cell Viability and Growth Assays
4.5. Invasion and Migration Assays
4.6. Co-Culture Assays
4.7. Flow Cytometry Analysis for Immune Subpopulations
4.8. Western Blot Analysis
4.9. RNA Extraction and Real-Time qPCR
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- National Cancer Institute. SEER Stat Fact Sheets: Lung and Bronchus. Available online: http://seer.cancer.gov/statfacts/html/lungb.html (accessed on 9 June 2016).
- American Cancer Society. Cancer Facts & Figures 2015. Available online: http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2015/ (accessed on 9 June 2016).
- Davis, R.J.; Van Waes, C.; Allen, C.T. Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and Tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol. 2016, 58, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.A.; Drake, V.; Huang, H.-S.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze T cells. OncoImmunology 2015, 4, e1016700. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int. Immunol. 2016, 28, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti-PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Page, D.B.; Postow, M.A.; Callahan, M.K.; Allison, J.P.; Wolchok, J.D. Immune Modulation in Cancer with Antibodies. Annu. Rev. Med. 2014, 65, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Romero, D. PD-1 says goodbye, TIM-3 says hello. Nat. Rev. Clin. Oncol. 2016, 13, 203. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasić, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, D.A.; Johnson, B.E.; Amler, L.C.; Goddard, A.D.; Heldens, S.L.; Herbst, R.S.; Ince, W.L.; Jänne, P.A.; Januario, T.; Klein, P.; et al. Mutations in the Epidermal Growth Factor Receptor and in KRAS Are Predictive and Prognostic Indicators in Patients with Non-Small-Cell Lung Cancer Treated With Chemotherapy Alone and in Combination With Erlotinib. J. Clin. Oncol. 2005, 23, 5900–5909. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS—Mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.A.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A.; et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genome Res. 2009, 23, 2140–2151. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Zheng, S.; Berel, D.; Yang, Y.; Raso, M.G.; Liu, D.D.; Wistuba, I.I.; Lozano, G.; et al. Expression Signatures of Metastatic Capacity in a Genetic Mouse Model of Lung Adenocarcinoma. PLoS ONE 2009, 4, e5401. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumor cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef]
- Peng, D.H.; Kundu, S.T.; Fradette, J.J.; Diao, L.; Tong, P.; Byers, L.A.; Wang, J.; Canales, J.R.; Villalobos, P.A.; Mino, B.; et al. ZEB1 suppression sensitizes KRAS mutant cancers to MEK inhibition by an IL17RD-dependent mechanism. Sci. Transl. Med. 2019, 11, eaaq1238. [Google Scholar] [CrossRef] [PubMed]
- Ungewiss, C.; Rizvi, Z.H.; Roybal, J.D.; Peng, D.H.; Gold, K.A.; Shin, D.-H.; Creighton, C.J.; Gibbons, D.L. The microRNA-200/Zeb1 axis regulates ECM-dependent β1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci. Rep. 2016, 6, 18652. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.H.; Ungewiss, C.; Tong, P.; Byers, L.A.; Wang, J.; Canales, J.R.; Villalobos, P.A.; Uraoka, N.; Mino, B.; Behrens, C.; et al. ZEB1 Induces LOXL2-Mediated Collagen Stabilization and Deposition in the Extracellular Matrix to Drive Lung Cancer Invasion and Metastasis. Oncogene 2016, 36, 1925–1938. [Google Scholar] [CrossRef] [PubMed]
- Carugo, A.; Genovese, G.; Seth, S.; Nezi, L.; Rose, J.L.; Bossi, D.; Cicalese, A.; Shah, P.K.; Viale, A.; Pettazzoni, P.F.; et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016, 16, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Winslow, M.M.; Dayton, T.L.; Verhaak, R.G.W.; Kim-Kiselak, C.; Snyder, E.L.; Feldser, D.M.; Hubbard, D.D.; DuPage, M.J.; Whittaker, C.A.; Hoersch, S.; et al. Suppression of Lung Adenocarcinoma Progression by Nkx2-1. Nature 2011, 473, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; García-Diaz, Á.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.P.; Cheung, Z.H.; Ip, N.Y. STAT3 as a Downstream Mediator of Trk Signaling and Functions. J. Boil. Chem. 2006, 281, 15636–15644. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef]
- Farago, A.F.; Le, L.P.; Zheng, Z.; Muzikansky, A.; Drilon, A.; Patel, M.; Bauer, T.M.; Liu, S.V.; Ou, S.-H.I.; Jackman, D.; et al. Durable Clinical Response to Entrectinib in NTRK1-Rearranged Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2015, 10, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, R.A.; Bivona, T.G. Tracking down response and resistance to TRK inhibitors. Cancer Discov. 2016, 6, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and drug sensitive NTRK1 rearrangements in lung cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- TRK Inhibitor Shows Early Promise. Cancer Discov. 2015, 6. [CrossRef]
- Yano, H.; Cong, F.; Birge, R.B.; Goff, S.P.; Chao, M.V. Association of the Abl tyrosine kinase with the Trk nerve growth factor receptor. J. Neurosci. Res. 2000, 59, 356–364. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konen, J.M.; Rodriguez, B.L.; Fradette, J.J.; Gibson, L.; Davis, D.; Minelli, R.; Peoples, M.D.; Kovacs, J.; Carugo, A.; Bristow, C.; et al. Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/p53 Mutant Lung Cancer. Cancers 2019, 11, 462. https://doi.org/10.3390/cancers11040462
Konen JM, Rodriguez BL, Fradette JJ, Gibson L, Davis D, Minelli R, Peoples MD, Kovacs J, Carugo A, Bristow C, et al. Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/p53 Mutant Lung Cancer. Cancers. 2019; 11(4):462. https://doi.org/10.3390/cancers11040462
Chicago/Turabian StyleKonen, Jessica M., B. Leticia Rodriguez, Jared J. Fradette, Laura Gibson, Denali Davis, Rosalba Minelli, Michael D. Peoples, Jeffrey Kovacs, Alessandro Carugo, Christopher Bristow, and et al. 2019. "Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/p53 Mutant Lung Cancer" Cancers 11, no. 4: 462. https://doi.org/10.3390/cancers11040462
APA StyleKonen, J. M., Rodriguez, B. L., Fradette, J. J., Gibson, L., Davis, D., Minelli, R., Peoples, M. D., Kovacs, J., Carugo, A., Bristow, C., Heffernan, T., & Gibbons, D. L. (2019). Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/p53 Mutant Lung Cancer. Cancers, 11(4), 462. https://doi.org/10.3390/cancers11040462