Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy

 , , , and add
Show full author list
, , , and add
Show full author list
{kind=link}
Abstract
1. Rethinking Cancer Therapy Development
2. Tumors Responsive to Immunotherapy
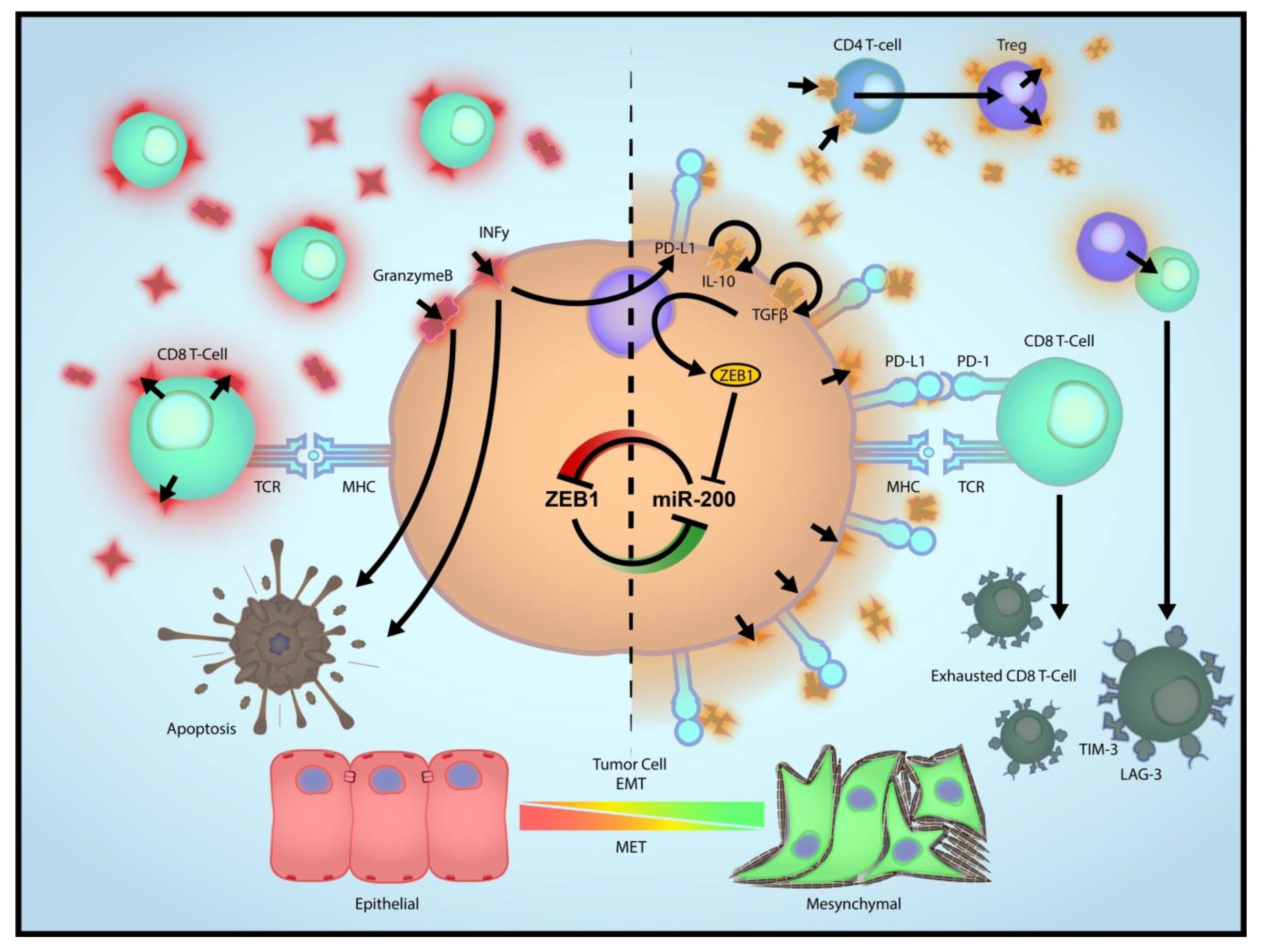
3. Role of EMT in Immune Evasion
4. EMT as a Driver of Immune Escape from ICBT
5. Relationship between CD8 T Cells and EMT, and Impact of EMT on ICBT
6. Impact of Immunosuppressive TME on ICBT
7. Role of Bioinformatics
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Casasent, A.K.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, N.E. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018, 172, 205–217.e212. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Wilkerson, P.M.; Lambros, M.B.; Campion-Flora, A.; Rodrigues, D.N.; Gauthier, A.; Cabral, C.; Pawar, V.; Mackay, A.; A’Hern, R.; et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J. Pathol. 2012, 227, 42–52. [Google Scholar] [CrossRef]
- Niyomnaitham, S.; Parinyanitikul, N.; Roothumnong, E.; Jinda, W.; Samarnthai, N.; Atikankul, T.; Suktitipat, B.; Thongnoppakhun, W.; Limwongse, C.; Pithukpakorn, M. Tumor mutational profile of triple negative breast cancer patients in Thailand revealed distinctive genetic alteration in chromatin remodeling gene. PeerJ 2019, 7, e6501. [Google Scholar] [CrossRef]
- Fardi, M.; Alivand, M.; Baradaran, B.; Farshdousti Hagh, M.; Solali, S. The crucial role of ZEB2: From development to epithelial-to-mesenchymal transition and cancer complexity. J. Cell Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liang, Y.; Li, S.; Zeng, F.; Meng, Y.; Chen, Z.; Liu, S.; Tao, Y.; Yu, F. The interplay of circulating tumor DNA and chromatin modification, therapeutic resistance, and metastasis. Mol. Cancer 2019, 18, 36. [Google Scholar] [CrossRef]
- Schwarzenbacher, D.; Balic, M.; Pichler, M. The role of microRNAs in breast cancer stem cells. Int. J. Mol. Sci. 2013, 14, 14712–14723. [Google Scholar] [CrossRef]
- Markiewicz, A.; Topa, J.; Nagel, A.; Skokowski, J.; Seroczynska, B.; Stokowy, T.; Welnicka-Jaskiewicz, M.; Zaczek, A.J. Spectrum of Epithelial-Mesenchymal Transition Phenotypes in Circulating Tumour Cells from Early Breast Cancer Patients. Cancers 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Andre, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef]
- Chouaib, S.; Janji, B.; Tittarelli, A.; Eggermont, A.; Thiery, J.P. Tumor plasticity interferes with anti-tumor immunity. Crit. Rev. Immunol. 2014, 34, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Terry, S.; Chouaib, S. EMT in immuno-resistance. Oncoscience 2015, 2, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, H.; Zhao, S.; Wang, Y.; Pu, H.; Wang, Y.; Zhang, Q. Expression of PD-L1 and prognosis in breast cancer: A meta-analysis. Oncotarget 2017, 8, 31347–31354. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab Monotherapy for Previously Treated Metastatic Triple-Negative Breast Cancer: Cohort A of the Phase 2 KEYNOTE-086 Study. Ann. Oncol. 2018. [Google Scholar] [CrossRef]
- Wang, Y.J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Langer, C.J.; Gadgeel, S.M.; Borghaei, H.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: A randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016, 17, 1497–1508. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Reck, M.; Rabe, K.F. Precision Diagnosis and Treatment for Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 849–861. [Google Scholar] [CrossRef]
- McArthur, H.L.; Page, D.B. Immunotherapy for the treatment of breast cancer: Checkpoint blockade, cancer vaccines, and future directions in combination immunotherapy. Clin. Adv. Hematol. Oncol. 2016, 14, 922–933. [Google Scholar] [PubMed]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M.; Galaine, J.; Guenat, D.; Mougey, V.; Perrard, J.; et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 2018, 7, e1423170. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhu, T.; Yue, Y.; Zhu, X.; Wang, J.; Liang, L. Preliminary mechanisms of regulating PDL1 expression in nonsmall cell lung cancer during the EMT process. Oncol. Rep. 2018, 40, 775–782. [Google Scholar] [CrossRef]
- Kumar, S.; Davra, V.; Obr, A.E.; Geng, K.; Wood, T.L.; De Lorenzo, M.S.; Birge, R.B. Crk adaptor protein promotes PD-L1 expression, EMT and immune evasion in a murine model of triple-negative breast cancer. Oncoimmunology 2017, 7, e1376155. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Koh, J.; Kim, M.Y.; Kwon, D.; Go, H.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H. PD-L1 expression is associated with epithelial-to-mesenchymal transition in adenocarcinoma of the lung. Hum. Pathol. 2016, 58, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Low-Marchelli, J.M.; Ardi, V.C.; Vizcarra, E.A.; van Rooijen, N.; Quigley, J.P.; Yang, J. Twist1 induces CCL2 and recruits macrophages to promote angiogenesis. Cancer Res. 2013, 73, 662–671. [Google Scholar] [CrossRef]
- Hsu, D.S.; Wang, H.J.; Tai, S.K.; Chou, C.H.; Hsieh, C.H.; Chiu, P.H.; Chen, N.J.; Yang, M.H. Acetylation of snail modulates the cytokinome of cancer cells to enhance the recruitment of macrophages. Cancer Cell 2014, 26, 534–548. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Otsuki, Y.; Saya, H.; Arima, Y. Prospects for new lung cancer treatments that target EMT signaling. Dev. Dyn. 2018, 247, 462–472. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; Laframboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Allison Stewart, C.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y.; et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017, 8, 28575–28587. [Google Scholar] [CrossRef]
- Lou, Y.; Diao, L.; Cuentas, E.R.; Denning, W.L.; Chen, L.; Fan, Y.H.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.A.; Behrens, C.; et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bae, J.S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediat. Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [PubMed]
- Carron, E.C.; Homra, S.; Rosenberg, J.; Coffelt, S.B.; Kittrell, F.; Zhang, Y.; Creighton, C.J.; Fuqua, S.A.; Medina, D.; Machado, H.L. Macrophages promote the progression of premalignant mammary lesions to invasive cancer. Oncotarget 2017, 8, 50731–50746. [Google Scholar] [CrossRef] [PubMed]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3117. [Google Scholar] [CrossRef]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, P.; Mudduluru, G.; Kumarswamy, R.; Rapa, I.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol. Cancer Res. 2010, 8, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Schwarz, J.K.; Lewis, J.S., Jr.; Huettner, P.C.; Rader, J.S.; Deasy, J.O.; Grigsby, P.W.; Wang, X. A microRNA expression signature for cervical cancer prognosis. Cancer Res. 2010, 70, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.A.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A.; et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009, 23, 2140–2151. [Google Scholar] [CrossRef]
- Ahn, Y.H.; Gibbons, D.L.; Chakravarti, D.; Creighton, C.J.; Rizvi, Z.H.; Adams, H.P.; Pertsemlidis, A.; Gregory, P.A.; Wright, J.A.; Goodall, G.J.; et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J. Clin. Investig. 2012, 122, 3170–3183. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ahn, Y.H.; Gibbons, D.L.; Zang, Y.; Lin, W.; Thilaganathan, N.; Alvarez, C.A.; Moreira, D.C.; Creighton, C.J.; Gregory, P.A.; et al. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J. Clin. Investig. 2011, 121, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Ungewiss, C.; Rizvi, Z.H.; Roybal, J.D.; Peng, D.H.; Gold, K.A.; Shin, D.H.; Creighton, C.J.; Gibbons, D.L. The microRNA-200/Zeb1 axis regulates ECM-dependent beta1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci. Rep. 2016, 6, 18652. [Google Scholar] [CrossRef] [PubMed]
- Schliekelman, M.J.; Gibbons, D.L.; Faca, V.M.; Creighton, C.J.; Rizvi, Z.H.; Zhang, Q.; Wong, C.H.; Wang, H.; Ungewiss, C.; Ahn, Y.H.; et al. Targets of the tumor suppressor miR-200 in regulation of the epithelial-mesenchymal transition in cancer. Cancer Res. 2011, 71, 7670–7682. [Google Scholar] [CrossRef]
- Mahmoud, S.M.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.; Ellis, I.O.; Green, A.R. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, E.; Gil, G.L.; Benito, A.C.; Gonzalez-Billalabeitia, E.; Conesa, M.A.; Garcia Garcia, T.; Garcia-Garre, E.; Vicente, V.; Ayala de la Pena, F. Tumor-infiltrating immune cell profiles and their change after neoadjuvant chemotherapy predict response and prognosis of breast cancer. Breast Cancer Res. 2014, 16, 488. [Google Scholar] [CrossRef]
- Iglesia, M.D.; Vincent, B.G.; Parker, J.S.; Hoadley, K.A.; Carey, L.A.; Perou, C.M.; Serody, J.S. Prognostic B-cell signatures using mRNA-seq in patients with subtype-specific breast and ovarian cancer. Clin. Cancer Res. 2014, 20, 3818–3829. [Google Scholar] [CrossRef]
- Liu, S.; Lachapelle, J.; Leung, S.; Gao, D.; Foulkes, W.D.; Nielsen, T.O. CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res. 2012, 14, R48. [Google Scholar] [CrossRef]
- Hudis, C.A. Trastuzumab--mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Shurin, M.R. Dual role of immunomodulation by anticancer chemotherapy. Nat. Med. 2013, 19, 20–22. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.J.; Kroemer, G. Mechanism of action of conventional and targeted anticancer therapies: Reinstating immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef]
- Emens, L.A.; Kok, M.; Ojalvo, L.S. Targeting the programmed cell death-1 pathway in breast and ovarian cancer. Curr. Opin. Obstet. Gynecol. 2016, 28, 142–147. [Google Scholar] [CrossRef]
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Savas, P.; Salgado, R.; Denkert, C.; Sotiriou, C.; Darcy, P.K.; Smyth, M.J.; Loi, S. Clinical relevance of host immunity in breast cancer: From TILs to the clinic. Nat. Rev. Clin. Oncol. 2016, 13, 228–241. [Google Scholar] [CrossRef]
- Greifenberg, V.; Ribechini, E.; Rossner, S.; Lutz, M.B. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur. J. Immunol. 2009, 39, 2865–2876. [Google Scholar] [CrossRef]
- Andersen, M.H.; Schrama, D.; Thor Straten, P.; Becker, J.C. Cytotoxic T cells. J. Investig. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.E.; Forman, J. The role of CD8 T cells in innate immunity and in antigen non-specific protection. Curr. Opin. Immunol. 2006, 18, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Dine, J.; Gordon, R.; Shames, Y.; Kasler, M.K.; Barton-Burke, M. Immune Checkpoint Inhibitors: An Innovation in Immunotherapy for the Treatment and Management of Patients with Cancer. Asia Pac. J. Oncol. Nurs. 2017, 4, 127–135. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef]
- Jackson, S.R.; Yuan, J.; Teague, R.M. Targeting CD8+ T-cell tolerance for cancer immunotherapy. Immunotherapy 2014, 6, 833–852. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Knutson, K.L.; Lu, H.; Stone, B.; Reiman, J.M.; Behrens, M.D.; Prosperi, C.M.; Gad, E.A.; Smorlesi, A.; Disis, M.L. Immunoediting of cancers may lead to epithelial to mesenchymal transition. J. Immunol. 2006, 177, 1526–1533. [Google Scholar] [CrossRef]
- Mahic, M.; Henjum, K.; Yaqub, S.; Bjornbeth, B.A.; Torgersen, K.M.; Tasken, K.; Aandahl, E.M. Generation of highly suppressive adaptive CD8(+)CD25(+)FOXP3(+) regulatory T cells by continuous antigen stimulation. Eur. J. Immunol. 2008, 38, 640–646. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef]
- Caja, F.; Vannucci, L. TGFbeta: A player on multiple fronts in the tumor microenvironment. J. Immunotoxicol. 2015, 12, 300–307. [Google Scholar] [CrossRef]
- Mani, S.A.; Yang, J.; Brooks, M.; Schwaninger, G.; Zhou, A.; Miura, N.; Kutok, J.L.; Hartwell, K.; Richardson, A.L.; Weinberg, R.A. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2007, 104, 10069–10074. [Google Scholar] [CrossRef] [PubMed]
- Dye, E.S. The antimetastatic function of concomitant antitumor immunity. II. Evidence that the generation of Ly-1+2+ effector T cells temporarily causes the destruction of already disseminated tumor cells. J. Immunol. 1986, 136, 1510–1515. [Google Scholar] [PubMed]
- Luo, H.; Zeng, C.; Fang, C.; Seeruttun, S.R.; Lv, L.; Wang, W. A new strategy using ALDHhigh-CD8+T cells to inhibit tumorigenesis. PLoS ONE 2014, 9, e103193. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, L.; Cao, S.; Zhu, Y.; MEI, Q. Gene delivery of TIPE2 inhibits breast cancer development and metastasis via CD8+ T and NK cell-mediated antitumor responses. Mol. Immunol. 2017, 85, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hofmann, M.; Wang, Q.; Teng, L.; Chlewicki, L.K.; Pircher, H.; Mariuzza, R.A. Structure of natural killer cell receptor KLRG1 bound to E-cadherin reveals basis for MHC-independent missing self recognition. Immunity 2009, 31, 35–46. [Google Scholar] [CrossRef]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [PubMed]
- Chockley, P.J.; Chen, J.; Chen, G.; Beer, D.G.; Standiford, T.J.; Keshamouni, V.G. Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer. J. Clin. Investig. 2018, 128, 1384–1396. [Google Scholar] [CrossRef]
- Stovgaard, E.S.; Nielsen, D.; Hogdall, E.; Balslev, E. Triple negative breast cancer - prognostic role of immune-related factors: A systematic review. Acta Oncol. 2017. [Google Scholar] [CrossRef]
- Xu, C.; Fillmore, C.M.; Koyama, S.; Wu, H.; Zhao, Y.; Chen, Z.; Herter-Sprie, G.S.; Akbay, E.A.; Tchaicha, J.H.; Altabef, A.; et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 2014, 25, 590–604. [Google Scholar] [CrossRef]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef]
- Welte, T.; Kim, I.S.; Tian, L.; Gao, X.; Wang, H.; Li, J.; Holdman, X.B.; Herschkowitz, J.I.; Pond, A.; Xie, G.; et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol. 2016, 18, 632–644. [Google Scholar] [CrossRef]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Zhang, X.H.; Rosen, J.M. Repurposing Antiestrogens for Tumor Immunotherapy. Cancer Discov. 2017, 7, 17–19. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Chang, J.T.; Gatza, M.L.; Lucas, J.E.; Barry, W.T.; Vaughn, P.; Nevins, J.R. SIGNATURE: A workbench for gene expression signature analysis. BMC Bioinformatics 2011, 12, 443. [Google Scholar] [CrossRef]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; Andre, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Wheeler, D.L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. Ther. 2011, 11, 777–792. [Google Scholar] [CrossRef]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Cheng, W.Y.; Kandel, J.J.; Yamashiro, D.J.; Canoll, P.; Anastassiou, D. A multi-cancer mesenchymal transition gene expression signature is associated with prolonged time to recurrence in glioblastoma. PLoS ONE 2012, 7, e34705. [Google Scholar] [CrossRef]
- Sethi, S.; Macoska, J.; Chen, W.; Sarkar, F.H. Molecular signature of epithelial-mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am. J. Transl. Res. 2010, 3, 90–99. [Google Scholar]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef]
- Sugio, T.; Miyawaki, K.; Kato, K.; Sasaki, K.; Yamada, K.; Iqbal, J.; Miyamoto, T.; Ohshima, K.; Maeda, T.; Miyoshi, H.; et al. Microenvironmental immune cell signatures dictate clinical outcomes for PTCL-NOS. Blood Adv. 2018, 2, 2242–2252. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.R.; Provenzano, E.; Dawson, S.J.; Blows, F.M.; Liu, B.; Shah, M.; Earl, H.M.; Poole, C.J.; Hiller, L.; Dunn, J.A.; et al. Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Ann. Oncol. 2014, 25, 1536–1543. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Miremadi, A.; Pinder, S.E.; Ellis, I.O.; Caldas, C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007, 8, R157. [Google Scholar] [CrossRef]
- Rody, A.; Holtrich, U.; Pusztai, L.; Liedtke, C.; Gaetje, R.; Ruckhaeberle, E.; Solbach, C.; Hanker, L.; Ahr, A.; Metzler, D.; et al. T-cell metagene predicts a favorable prognosis in estrogen receptor-negative and HER2-positive breast cancers. Breast Cancer Res. 2009, 11, R15. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, M.L.; Kmieciak, M.; Idowu, M.O.; Manjili, R.; Zhao, Y.; Grimes, M.; Dumur, C.; Wang, E.; Ramakrishnan, V.; Wang, X.Y.; et al. A signature of immune function genes associated with recurrence-free survival in breast cancer patients. Breast Cancer Res. Treat. 2012, 131, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, M.L.; Idowu, M.O.; Zhao, Y.; Khalak, H.; Payne, K.K.; Wang, X.Y.; Dumur, C.I.; Bedognetti, D.; Tomei, S.; Ascierto, P.A.; et al. Molecular signatures mostly associated with NK cells are predictive of relapse free survival in breast cancer patients. J. Transl. Med. 2013, 11, 145. [Google Scholar] [CrossRef]
- Perez, E.A.; Thompson, E.A.; Ballman, K.V.; Anderson, S.K.; Asmann, Y.W.; Kalari, K.R.; Eckel-Passow, J.E.; Dueck, A.C.; Tenner, K.S.; Jen, J.; et al. Genomic analysis reveals that immune function genes are strongly linked to clinical outcome in the North Central Cancer Treatment Group n9831 Adjuvant Trastuzumab Trial. J. Clin. Oncol. 2015, 33, 701–708. [Google Scholar] [CrossRef]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Muller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef]
- Forero, A.; Li, Y.; Chen, D.; Grizzle, W.E.; Updike, K.L.; Merz, N.D.; Downs-Kelly, E.; Burwell, T.C.; Vaklavas, C.; Buchsbaum, D.J.; et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 390–399. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. https://doi.org/10.3390/cancers11050714
Soundararajan R, Fradette JJ, Konen JM, Moulder S, Zhang X, Gibbons DL, Varadarajan N, Wistuba II, Tripathy D, Bernatchez C, et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers. 2019; 11(5):714. https://doi.org/10.3390/cancers11050714
Chicago/Turabian StyleSoundararajan, Rama, Jared J. Fradette, Jessica M. Konen, Stacy Moulder, Xiang Zhang, Don L. Gibbons, Navin Varadarajan, Ignacio I. Wistuba, Debasish Tripathy, Chantale Bernatchez, and et al. 2019. "Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy" Cancers 11, no. 5: 714. https://doi.org/10.3390/cancers11050714
APA StyleSoundararajan, R., Fradette, J. J., Konen, J. M., Moulder, S., Zhang, X., Gibbons, D. L., Varadarajan, N., Wistuba, I. I., Tripathy, D., Bernatchez, C., Byers, L. A., Chang, J. T., Contreras, A., Lim, B., Parra, E. R., Roarty, E. B., Wang, J., Yang, F., Barton, M., ... Mani, S. A. (2019). Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers, 11(5), 714. https://doi.org/10.3390/cancers11050714