Targeted siRNA Nanoparticles for Mammary Carcinoma Therapy


Abstract
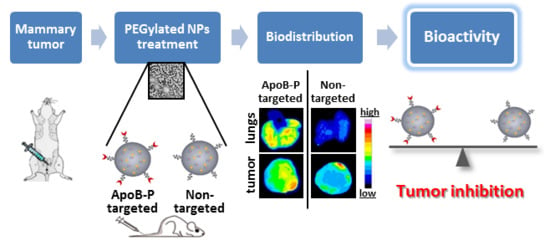
1. Introduction
2. Results
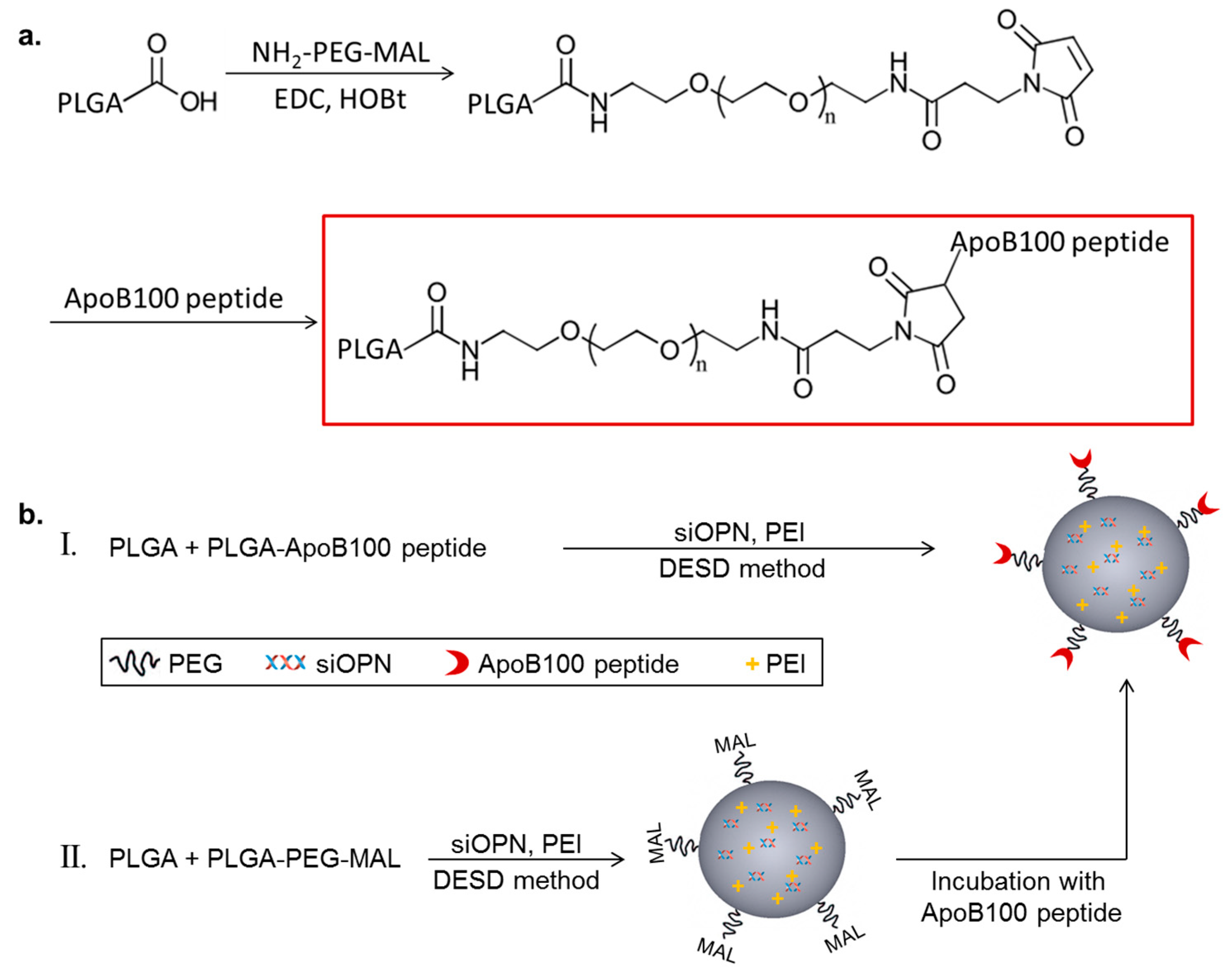
2.1. Poly(d,l-Lactic-Co-Glycolic Acid) Apolipoprotein B100 Peptide (PLGA-ApoB-P) Synthesis
2.2. Nanoparticles (NPs) Physicochemical Properties
2.3. In Vitro Binding and Uptake
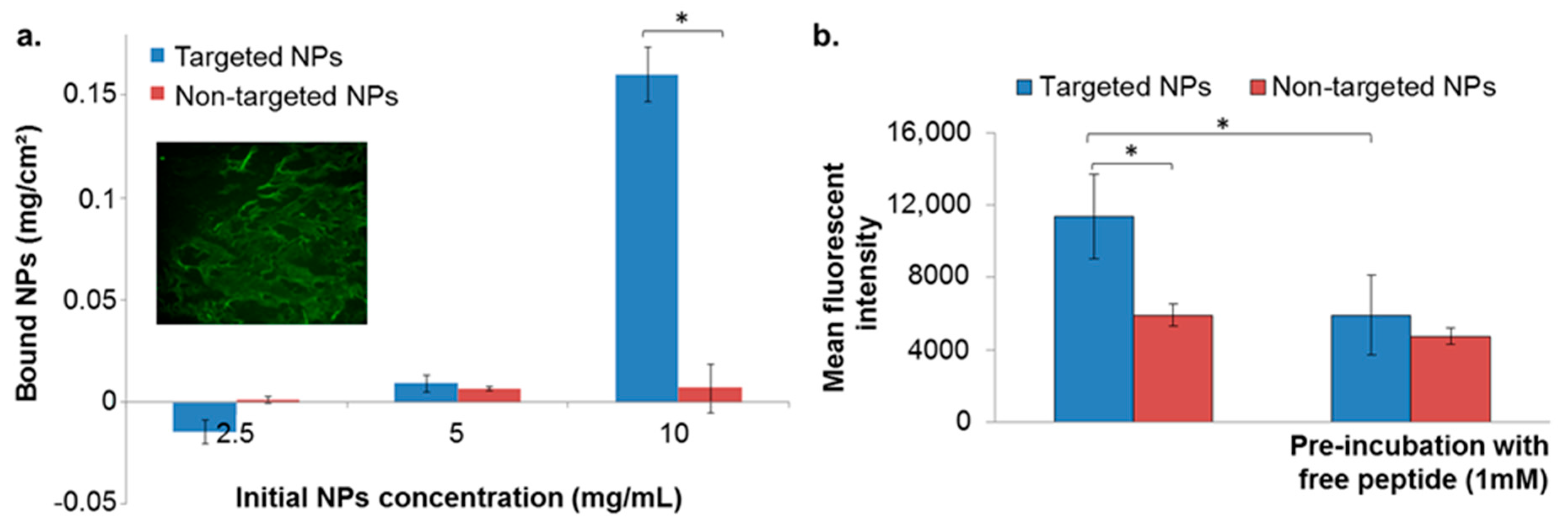
2.3.1. Binding to Isolated Extracellular Matrix (ECM)
2.3.2. Binding to the Basement Membrane (BM) Matrix
2.3.3. Uptake into the MDA-MB-231 Cell Line
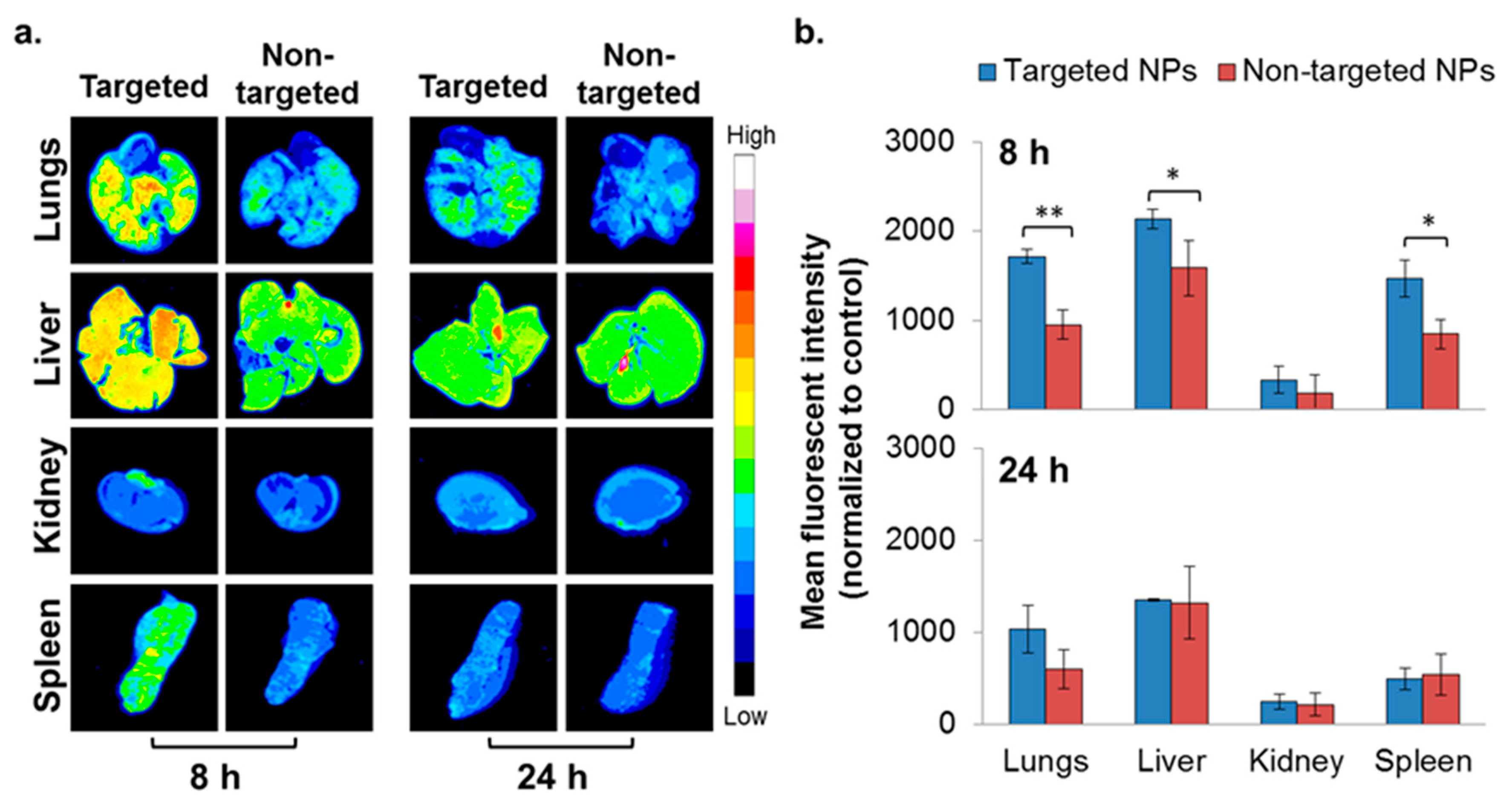
2.4. In Vivo Biodistribution—4T1 Intravenous (IV) Model
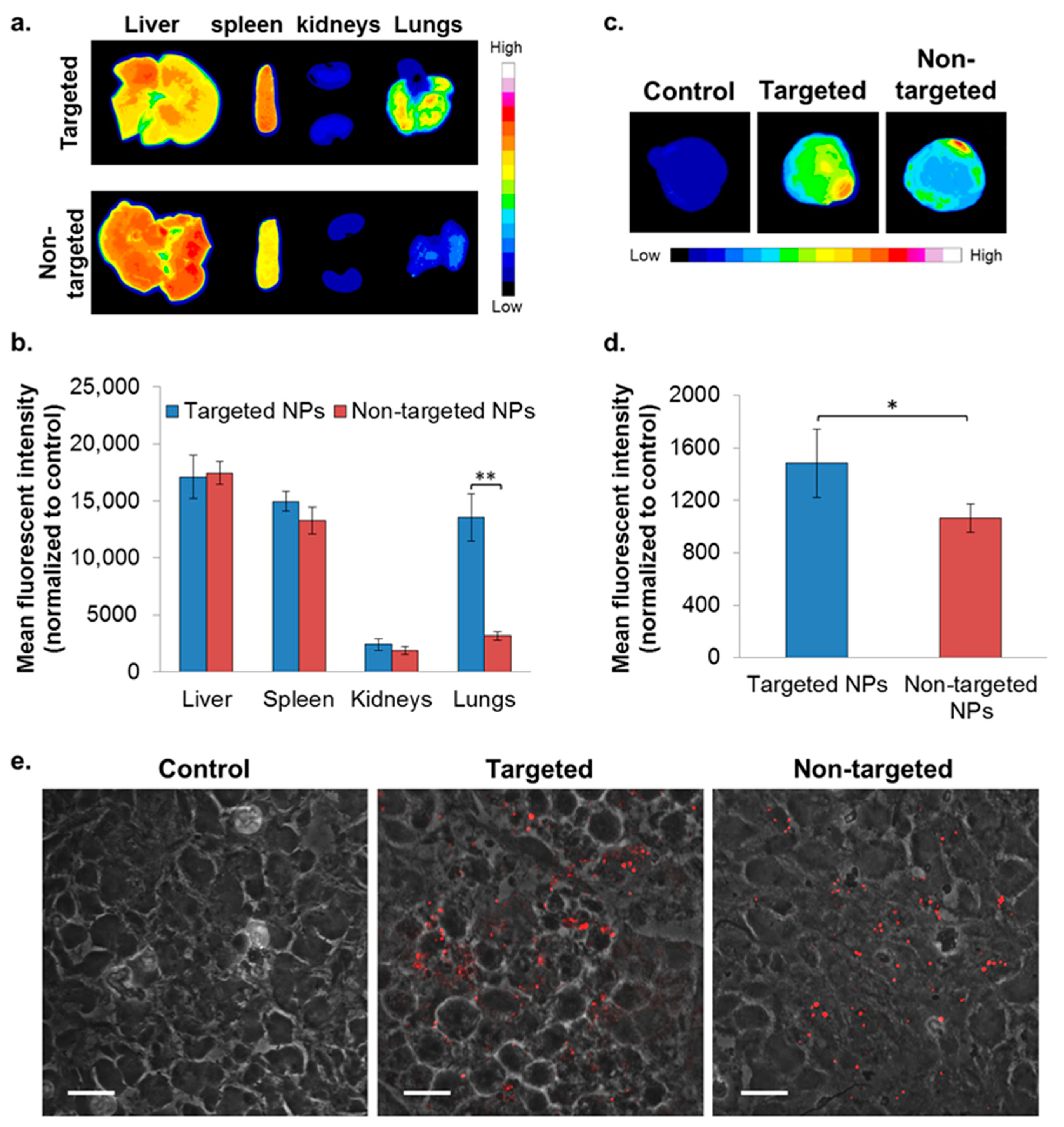
2.5. In Vivo Biodistribution—Orthotopic Xenograft Model
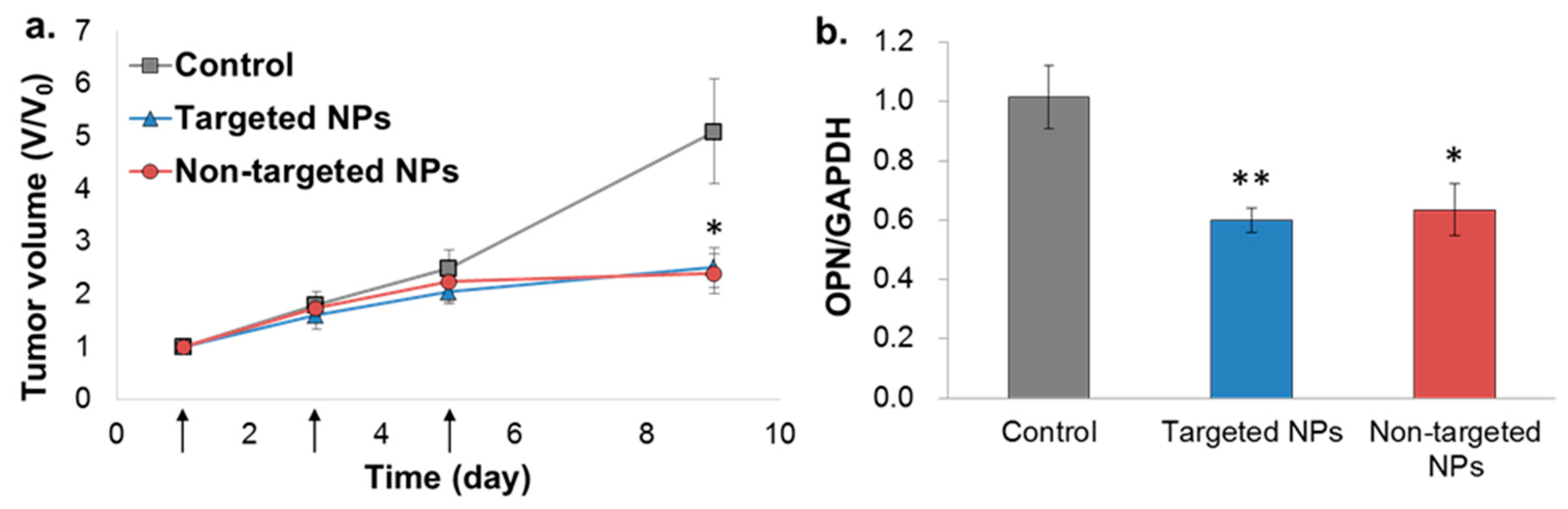
2.6. In Vivo Bioactivity—Orthotopic Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. PLGA-ApoB-P Synthesis
4.3. Nanoparticles Preparation
4.4. Determination of NP Size, Polydispersity, and Surface Charge
4.5. Determination of siOPN Content
4.6. In Vitro Binding Studies
4.6.1. NPs Binding to the Isolated ECM
4.6.2. NPs Binding to the BM Matrix
4.7. Cellular Uptake Studies
4.7.1. Quantification of NPs Cellular Uptake
4.7.2. Visualization of NPs Cellular Uptake
4.8. In Vivo Mice Models
4.8.1. Mammary Carcinoma 4T1 IV Model
4.8.2. Mammary Carcinoma Orthotopic Mice Model
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Sarisozen, C.; Pan, J.; Dutta, I.; Torchilin, V.P. Polymers in the co-delivery of siRNA and anticancer drugs to treat multidrug-resistant tumors. J. Pharm. Investig. 2017, 47, 37–49. [Google Scholar] [CrossRef]
- Mir, M.; Ahmed, N.; Rehman, A.U. Recent applications of PLGA based nanostructures in drug delivery. Colloids Surf. B Biointerfaces 2017, 159, 217–231. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Preat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Patil, Y.; Panyam, J. Polymeric nanoparticles for siRNA delivery and gene silencing. Int. J. Pharm. 2009, 367, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Levy, R.J.; Gao, J.; Fishbein, I.; Kousaev, V.; Sosnowski, S.; Slomkowski, S.; Golomb, G. Sustained delivery and expression of DNA encapsulated in polymeric nanoparticles. Gene Ther. 2000, 7, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Sacks, H.; Najajreh, Y.; Tchaikovski, V.; Gao, G.; Elazer, V.; Dahan, R.; Gati, I.; Kanaan, M.; Waltenberger, J.; Golomb, G. Novel PDGFbetaR antisense encapsulated in polymeric nanospheres for the treatment of restenosis. Gene Ther. 2002, 9, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Sah, H.; Thoma, L.A.; Desu, H.R.; Sah, E.; Wood, G.C. Concepts and practices used to develop functional PLGA-based nanoparticulate systems. Int. J. Nanomed. 2013, 8, 747–765. [Google Scholar] [CrossRef]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef]
- Barua, S.; Mitragotri, S. Challenges associated with Penetration of Nanoparticles across Cell and Tissue Barriers: A Review of Current Status and Future Prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Simberg, D. Nanoparticle transport pathways into tumors. J. Nanopart. Res. 2018, 20, 169. [Google Scholar] [CrossRef]
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C.; Andresen, T.L. Factors controlling nanoparticle pharmacokinetics: An integrated analysis and perspective. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [Google Scholar] [CrossRef] [PubMed]
- Anchordoquy, T.J.; Barenholz, Y.; Boraschi, D.; Chorny, M.; Decuzzi, P.; Dobrovolskaia, M.A.; Farhangrazi, Z.S.; Farrell, D.; Gabizon, A.; Ghandehari, H.; et al. Mechanisms and barriers in cancer nanomedicine: Addressing challenges, looking for solutions. ACS Nano 2017, 11, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sherifi, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 6315–6320. [Google Scholar] [CrossRef] [PubMed]
- Fasehee, H.; Dinarvand, R.; Ghavamzadeh, A.; Esfandyari-Manesh, M.; Moradian, H.; Faghihi, S.; Ghaffari, S.H. Delivery of disulfiram into breast cancer cells using folate-receptor-targeted PLGA-PEG nanoparticles: In vitro and in vivo investigations. J. Nanobiotechnol. 2016, 14, 32. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, H.; Yu, Y.; Chen, Y.; Wang, D.; Zhang, G.; Zhou, G.; Liu, J.; Sun, Z.; Sun, D.; et al. Biodegradable self-assembled nanoparticles of poly (d,l-lactide-co-glycolide)/hyaluronic acid block copolymers for target delivery of docetaxel to breast cancer. Biomaterials 2014, 35, 550–566. [Google Scholar] [CrossRef]
- Nicolas, J.; Mura, S.; Brambilla, D.; Mackiewicz, N.; Couvreur, P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem. Soc. Rev. 2013, 42, 1147–1235. [Google Scholar] [CrossRef]
- Elazar, V.; Adwan, H.; Rohekar, K.; Zepp, M.; Lifshitz-Shovali, R.; Berger, M.R.; Golomb, G. Biodistribution of antisense nanoparticles in mammary carcinoma rat model. Drug Deliv. 2010, 17, 408–418. [Google Scholar] [CrossRef]
- Ben David-Naim, M.; Grad, E.; Aizik, G.; Nordling-David, M.M.; Moshel, O.; Granot, Z.; Golomb, G. Polymeric nanoparticles of siRNA prepared by a double-emulsion solvent-diffusion technique: Physicochemical properties, toxicity, biodistribution and efficacy in a mammary carcinoma mice model. Biomaterials 2017, 145, 154–167. [Google Scholar] [CrossRef]
- Reufsteck, C.; Lifshitz-Shovali, R.; Zepp, M.; Bauerle, T.; Kubler, D.; Golomb, G.; Berger, M.R. Silencing of skeletal metastasis-associated genes impairs migration of breast cancer cells and reduces osteolytic bone lesions. Clin. Exp. Metastasis 2012, 29, 441–456. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Tsolakis, I.; Tzanakakis, G.N.; Karamanos, N.K. Chondroitin sulfate as a key molecule in the development of atherosclerosis and cancer progression. Adv. Pharmacol. 2006, 53, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Rensen, P.C.; de Vrueh, R.L.; Kuiper, J.; Bijsterbosch, M.K.; Biessen, E.A.; van Berkel, T.J. Recombinant lipoproteins: Lipoprotein-like lipid particles for drug targeting. Adv. Drug Deliv. Rev. 2001, 47, 251–276. [Google Scholar] [CrossRef]
- Insua-Rodriguez, J.; Oskarsson, T. The extracellular matrix in breast cancer. Adv. Drug Deliv. Rev. 2016, 97, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Knott, T.J.; Pease, R.J.; Powell, L.M.; Wallis, S.C.; Rall, S.C., Jr.; Innerarity, T.L.; Blackhart, B.; Taylor, W.H.; Marcel, Y.; Milne, R.; et al. Complete protein sequence and identification of structural domains of human apolipoprotein B. Nature 1986, 323, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Weisgraber, K.H.; Rall, S.C., Jr. Human apolipoprotein B-100 heparin-binding sites. J. Biol. Chem. 1987, 262, 11097–11103. [Google Scholar] [PubMed]
- Camejo, G.; Hurt-Camejo, E.; Wiklund, O.; Bondjers, G. Association of apo B lipoproteins with arterial proteoglycans: Pathological significance and molecular basis. Atherosclerosis 1998, 139, 205–222. [Google Scholar] [CrossRef]
- Golomb, G.; Sacks, H.; Najajreh, Y.; Fishbein, I.; Chorny, M. Nanoparticles Containing Polymeric nucleic Acid Homologs. U.S. Patent US8178128B2, 15 May 2012. [Google Scholar]
- Bellahcene, A.; Castronovo, V.; Ogbureke, K.U.; Fisher, L.W.; Fedarko, N.S. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): Multifunctional proteins in cancer. Nat. Rev. Cancer 2008, 8, 212–226. [Google Scholar] [CrossRef]
- Wai, P.Y.; Kuo, P.C. Osteopontin: Regulation in tumor metastasis. Cancer Metastasis Rev. 2008, 27, 103–118. [Google Scholar] [CrossRef]
- Shevde, L.A.; Samant, R.S. Role of osteopontin in the pathophysiology of cancer. Matrix Biol. 2014, 37, 131–141. [Google Scholar] [CrossRef]
- Bramwell, V.H.; Doig, G.S.; Tuck, A.B.; Wilson, S.M.; Tonkin, K.S.; Tomiak, A.; Perera, F.; Vandenberg, T.A.; Chambers, A.F. Serial plasma osteopontin levels have prognostic value in metastatic breast cancer. Clin. Cancer Res. 2006, 12, 3337–3343. [Google Scholar] [CrossRef]
- Adwan, H.; Bauerle, T.; Najajreh, Y.; Elazer, V.; Golomb, G.; Berger, M.R. Decreased levels of osteopontin and bone sialoprotein II are correlated with reduced proliferation, colony formation, and migration of GFP-MDA-MB-231 cells. Int. J. Oncol. 2004, 24, 1235–1244. [Google Scholar] [CrossRef]
- Elazar, V.; Adwan, H.; Bauerle, T.; Rohekar, K.; Golomb, G.; Berger, M.R. Sustained delivery and efficacy of polymeric nanoparticles containing osteopontin and bone sialoprotein antisenses in rats with breast cancer bone metastasis. Int. J. Cancer 2010, 126, 1749–1760. [Google Scholar] [CrossRef]
- Cho, W.Y.; Hong, S.H.; Singh, B.; Islam, M.A.; Lee, S.; Lee, A.Y.; Gankhuyag, N.; Kim, J.E.; Yu, K.N.; Kim, K.H.; et al. Suppression of tumor growth in lung cancer xenograft model mice by poly(sorbitol-co-PEI)-mediated delivery of osteopontin siRNA. Eur. J. Pharm. Biopharm. 2015, 94, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Rashid, O.M.; Nagahashi, M.; Ramachandran, S.; Dumur, C.I.; Schaum, J.C.; Yamada, A.; Aoyagi, T.; Milstien, S.; Spiegel, S.; Takabe, K. Is tail vein injection a relevant breast cancer lung metastasis model? J. Thorac. Dis. 2013, 5, 385–392. [Google Scholar] [CrossRef]
- Woodrow, K.A.; Cu, Y.; Booth, C.J.; Saucier-Sawyer, J.K.; Wood, M.J.; Saltzman, W.M. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat. Mater. 2009, 8, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Cuong, N.V.; Jiang, J.L.; Li, Y.L.; Chen, J.R.; Jwo, S.C.; Hsieh, M.F. Doxorubicin-loaded PEG-PCL-PEG micelle using xenograft model of nude mice: Effect of multiple administration of micelle on the suppression of human breast cancer. Cancers 2010, 3, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Tong, R.; Langer, R. Nanomedicines targeting the tumor microenvironment. Cancer J. 2015, 21, 314–321. [Google Scholar] [CrossRef]
- Ahmadzada, T.; Reid, G.; McKenzie, D.R. Fundamentals of siRNA and miRNA therapeutics and a review of targeted nanoparticle delivery systems in breast cancer. Biophys. Rev. 2018, 10, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Innerarity, T.L.; Weisgraber, K.H.; Rall, S.C., Jr.; Mahley, R.W. Functional domains of apolipoprotein E and apolipoprotein B. Acta Med. Scand. Suppl. 1987, 715, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Olsson, U.; Camejo, G.; Hurt-Camejo, E.; Elfsber, K.; Wiklund, O.; Bondjers, G. Possible functional interactions of apolipoprotein B-100 segments that associate with cell proteoglycans and the ApoB/E receptor. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Lee, I.; Zhu, W.; Arnold, K.; Taylor, S.; Innerarity, T.L. Identification of the low density lipoprotein receptor-binding site in apolipoprotein B100 and the modulation of its binding activity by the carboxyl terminus in familial defective apo-B100. J. Clin. Investig. 1998, 101, 1084–1093. [Google Scholar] [CrossRef]
- Olsson, U.; Camejo, G.; Olofsson, S.O.; Bondjers, G. Molecular parameters that control the association of low density lipoprotein apo B-100 with chondroitin sulphate. Biochim. Biophys. Acta 1991, 1097, 37–44. [Google Scholar] [CrossRef]
- Milne, R.; Theolis, R., Jr.; Maurice, R.; Pease, R.J.; Weech, P.K.; Rassart, E.; Fruchart, J.C.; Scott, J.; Marcel, Y.L. The use of monoclonal antibodies to localize the low density lipoprotein receptor-binding domain of apolipoprotein B. J. Biol. Chem. 1989, 264, 19754–19760. [Google Scholar] [PubMed]
- Grayson, A.C.; Doody, A.M.; Putnam, D. Biophysical and structural characterization of polyethylenimine-mediated siRNA delivery in vitro. Pharm. Res. 2006, 23, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Antalis, C.J.; Uchida, A.; Buhman, K.K.; Siddiqui, R.A. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin. Exp. Metastasis 2011, 28, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Koo, C.Y.; Sen, Y.P.; Bay, B.H.; Yip, G.W. Targeting heparan sulfate proteoglycans in breast cancer treatment. Recent Pat. Anticancer Drug Discov. 2008, 3, 151–158. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Yip, G.W.; Stock, C.; Pan, J.W.; Neubauer, C.; Poeter, M.; Pupjalis, D.; Koo, C.Y.; Kelsch, R.; Schule, R.; et al. Targeting of syndecan-1 by microRNA miR-10b promotes breast cancer cell motility and invasiveness via a Rho-GTPase- and E-cadherin-dependent mechanism. Int. J. Cancer 2012, 131, E884–E896. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Peterse, J.L.; van’t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Zhang, H.; Wong, C.C.; Wei, H.; Gilkes, D.M.; Korangath, P.; Chaturvedi, P.; Schito, L.; Chen, J.; Krishnamachary, B.; Winnard, P.T., Jr.; et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2012, 31, 1757–1770. [Google Scholar] [CrossRef]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef]
- Padua, D.; Zhang, X.H.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massague, J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Nakamura, K.; Iwai, S.; Murakami, M.; Itoh, T.; Kijima, H.; Shipley, J.M.; Senior, R.M.; Shibuya, M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2002, 2, 289–300. [Google Scholar] [CrossRef]
- Burns, A.R.; Smith, C.W.; Walker, D.C. Unique structural features that influence neutrophil emigration into the lung. Physiol. Rev. 2003, 83, 309–336. [Google Scholar] [CrossRef]
- Uyechi, L.S.; Gagne, L.; Thurston, G.; Szoka, F.C., Jr. Mechanism of lipoplex gene delivery in mouse lung: Binding and internalization of fluorescent lipid and DNA components. Gene Ther. 2001, 8, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; von Roemeling, C.A.; Chen, Y.; Qie, Y.; Liu, X.; Chen, J.; Kim, B.Y.S. Designing nanomedicine for immuno-oncology. Nat. Biomed. Eng. 2017, 1, 0029. [Google Scholar] [CrossRef]
- Rodero, M.P.; Poupel, L.; Loyher, P.L.; Hamon, P.; Licata, F.; Pessel, C.; Hume, D.A.; Combadiere, C.; Boissonnas, A. Immune surveillance of the lung by migrating tissue monocytes. eLife 2015, 4, e07847. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Owens, D.E., 3rd; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Natfji, A.A.; Ravishankar, D.; Osborn, H.M.I.; Greco, F. Parameters Affecting the Enhanced Permeability and Retention Effect: The Need for Patient Selection. J. Pharm. Sci. 2017, 106, 3179–3187. [Google Scholar] [CrossRef]
- Maeda, H. Polymer therapeutics and the EPR effect. J. Drug Target. 2017, 25, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.L.; Reuter, K.G.; Luft, J.C.; Pecot, C.V.; Zamboni, W.; DeSimone, J.M. Mediating Passive Tumor Accumulation through Particle Size, Tumor Type, and Location. Nano Lett. 2017, 17, 2879–2886. [Google Scholar] [CrossRef] [PubMed]
- Sykes, E.A.; Chen, J.; Zheng, G.; Chan, W.C. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano 2014, 8, 5696–5706. [Google Scholar] [CrossRef]
- Park, K. Facing the truth about nanotechnology in drug delivery. ACS Nano 2013, 7, 7442–7447. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, W.; Zhu, G.; Xie, J.; Chen, X. Rethinking cancer nanotheranostics. Nat. Rev. Mater. 2017, 2, 17024. [Google Scholar] [CrossRef]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Farhangrazi, Z.S. Just so stories: The random acts of anti-cancer nanomedicine performance. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug Delivery: Is the Enhanced Permeability and Retention Effect Sufficient for Curing Cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Dai, Q.; Wilhelm, S.; Ding, D.; Syed, A.M.; Sindhwani, S.; Zhang, Y.; Chen, Y.Y.; MacMillan, P.; Chan, W.C.W. Quantifying the ligand-coated nanoparticle delivery to cancer cells in solid tumors. ACS Nano 2018, 12, 8423–8435. [Google Scholar] [CrossRef]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The thiol-michael addition click reaction: A powerful and widely used tool in materials chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Wong, M.K.; Gotlieb, A.I. In vitro reendothelialization of a single-cell wound. Role of microfilament bundles in rapid lamellipodia-mediated wound closure. Lab. Investig. 1984, 51, 75–81. [Google Scholar]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NP Type | Size (d., nm) | Polydispersity Index (PDI) | Surface Charge (ζ Potential, mV) | siOPN Loading (µg/mg NPs) | Encapsulation Yield (%) |
|---|---|---|---|---|---|
| Targeted empty | 212.4 ± 3.6 | 0.21 ± 0.02 | −0.9 ± 0.2 | - | - |
| Non-targeted empty | 207.3 ± 4.3 | 0.17 ± 0.02 | −1.1 ± 0.1 | - | - |
| Targeted siOPN | 233.9 ± 3.7 | 0.29 ± 0.01 | −0.6 ± 0.2 | 4.9 ± 0.2 | 29.6 ± 0.9 |
| Non-targeted siOPN | 228.5 ± 23.0 | 0.31 ± 0.04 | −0.5 ± 0.1 | 5.1 ± 0.2 | 31.0 ± 1.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben-David-Naim, M.; Dagan, A.; Grad, E.; Aizik, G.; Nordling-David, M.M.; Morss Clyne, A.; Granot, Z.; Golomb, G. Targeted siRNA Nanoparticles for Mammary Carcinoma Therapy. Cancers 2019, 11, 442. https://doi.org/10.3390/cancers11040442
Ben-David-Naim M, Dagan A, Grad E, Aizik G, Nordling-David MM, Morss Clyne A, Granot Z, Golomb G. Targeted siRNA Nanoparticles for Mammary Carcinoma Therapy. Cancers. 2019; 11(4):442. https://doi.org/10.3390/cancers11040442
Chicago/Turabian StyleBen-David-Naim, Meital, Arie Dagan, Etty Grad, Gil Aizik, Mirjam M. Nordling-David, Alisa Morss Clyne, Zvi Granot, and Gershon Golomb. 2019. "Targeted siRNA Nanoparticles for Mammary Carcinoma Therapy" Cancers 11, no. 4: 442. https://doi.org/10.3390/cancers11040442
APA StyleBen-David-Naim, M., Dagan, A., Grad, E., Aizik, G., Nordling-David, M. M., Morss Clyne, A., Granot, Z., & Golomb, G. (2019). Targeted siRNA Nanoparticles for Mammary Carcinoma Therapy. Cancers, 11(4), 442. https://doi.org/10.3390/cancers11040442