A Simple and Highly Specific MassARRAY-Based Stool DNA Assay to Prioritize Follow-up Decisions in Fecal Immunochemical Test-Positive Individuals


 ,
,
Abstract
1. Introduction
2. Results
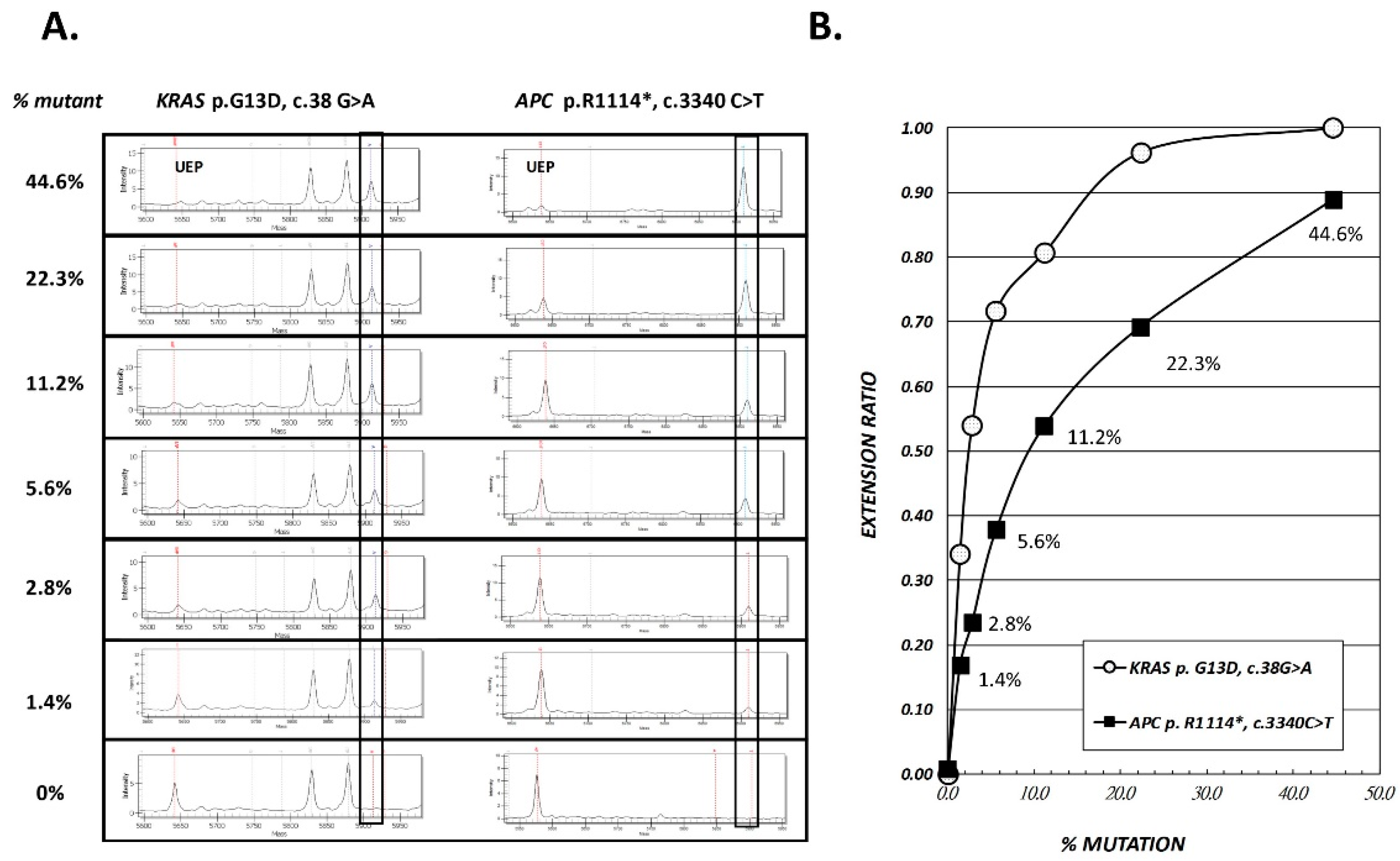
2.1. Accuracy and Analytical Sensitivity of SABER MassARRAY Assay, as Evaluated in LoVo Cell Line and Tissue DNA
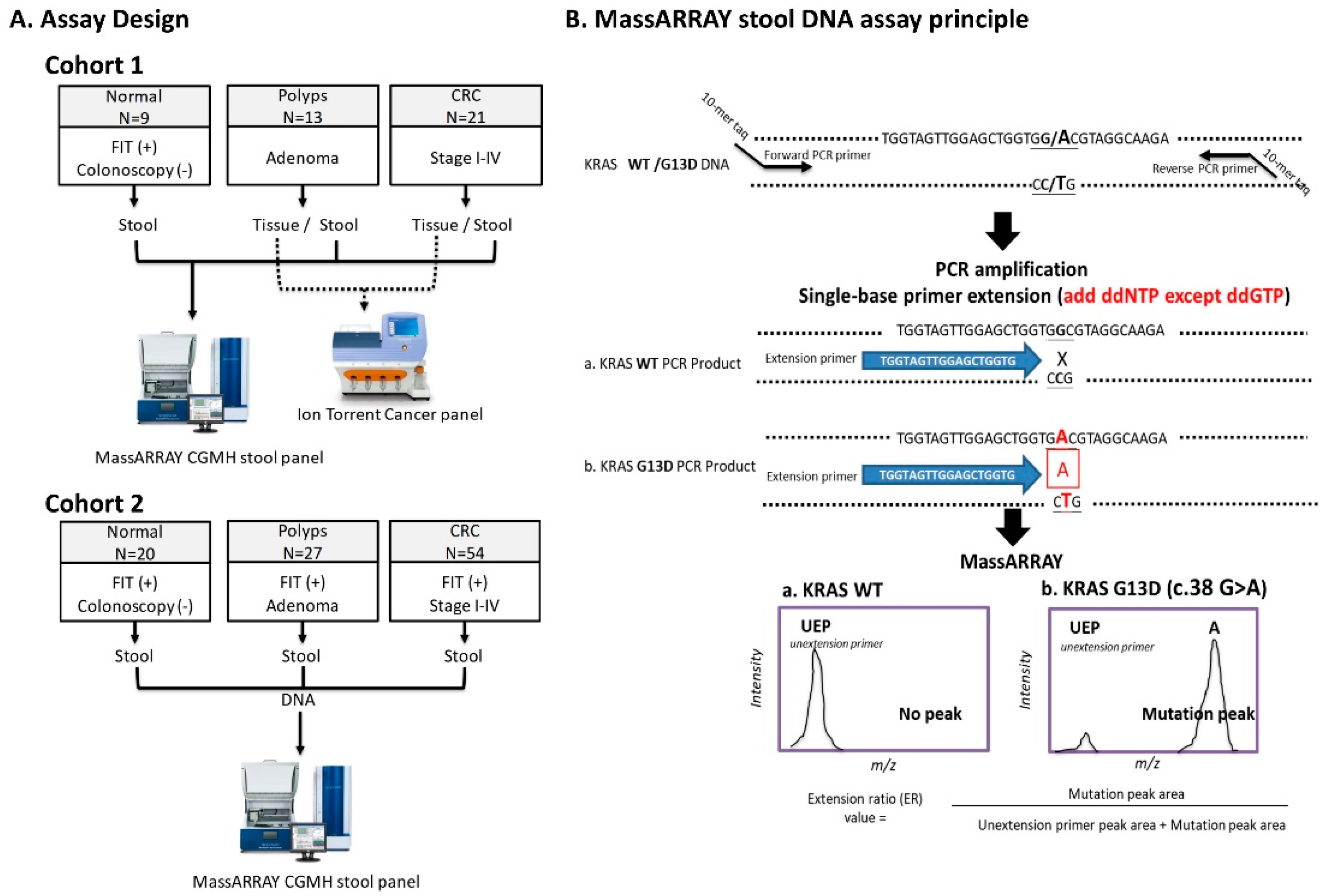
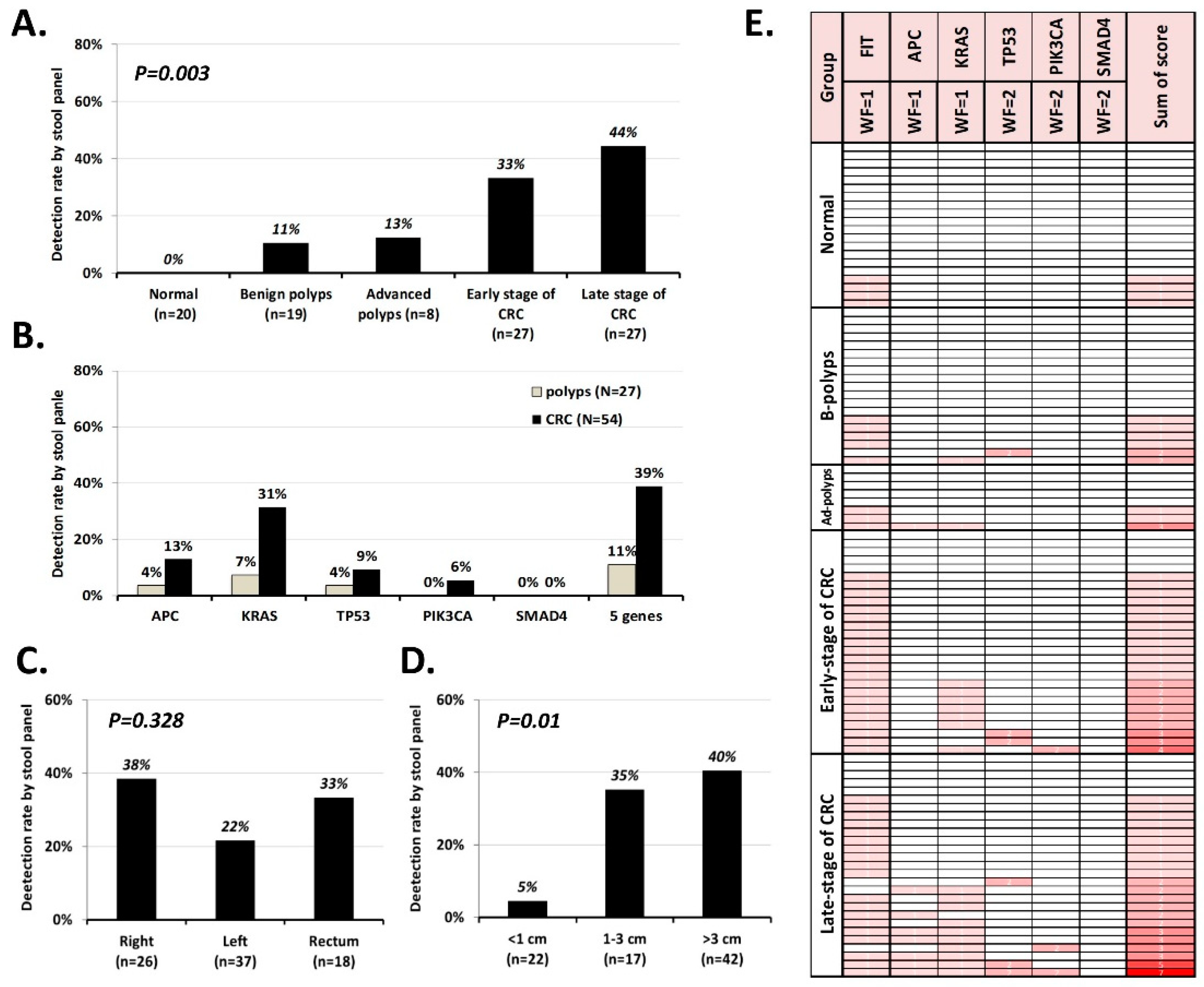
2.2. Performance of MassARRAY Stool DNA Panel in Cohort 1 Samples and Concordance of Mutations Detected in Stool and Matched Tissue Samples
2.3. Validating the Performance of the sDNA Panel in Cohort 2
3. Discussion
4. Materials and Methods
4.1. Selecting Genes and Variants for the sDNA Panel
4.2. LoVo Cell Line
4.3. Assay Design and Study Subjects
4.4. Extraction of DNA from Stool Samples
4.5. RNaseP Real-time PCR for Quantitation of Human DNA
4.6. Establishment of a MassARRAY-based Method for Detecting Rare Mutations
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chen, Y.-Y.; Chen, T.-H.; Su, M.-Y.; Ning, H.-C.; Kuo, C.-J.; Lin, W.-P.; Ho, Y.-P.; Lin, C.-J.; Hsu, C.-M.; Chiu, C.-T.; et al. Accuracy of immunochemical fecal occult blood test for detecting colorectal neoplasms in individuals undergoing health check-ups. Adv. Dig. Med. 2014, 1, 74–79. [Google Scholar] [CrossRef]
- Burt, R.W. Colon cancer screening. Gastroenterology 2000, 119, 837–853. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.W.; Chen, H.H.; Wu, M.S.; Chiu, H.M. Taiwanese Nationwide Colorectal Cancer Screening Program. Current status and future challenge of population-based organized colorectal cancer screening: Lesson from the first decade of Taiwanese program. J. Formos. Med. Assoc. 2018, 117, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Wang, A.Q.; Chen, Q.Q. DNA methylation assay for colorectal carcinoma. Cancer Biol. Med. 2017, 14, 42–49. [Google Scholar] [PubMed]
- Ahlquist, D.A. Molecular detection of colorectal neoplasia. Gastroenterology 2010, 138, 2127–2139. [Google Scholar] [CrossRef]
- Davies, R.J.; Miller, R.; Coleman, N. Colorectal cancer screening: Prospects for molecular stool analysis. Nat. Rev. Cancer 2005, 5, 199–209. [Google Scholar] [CrossRef]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef]
- Cotter, T.G.; Burger, K.N.; Devens, M.E.; Simonson, J.A.; Lowrie, K.L.; Heigh, R.I.; Mahoney, D.W.; Johnson, D.H.; Ahlquist, D.A.; Kisiel, J.B. Long-term Follow-up of Patients Having False-Positive Multitarget Stool DNA Tests after Negative Screening Colonoscopy: The LONG-HAUL Cohort Study. Cancer Epidemiol. Biomark. Prev. 2017, 26, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Levin, T.R.; Corley, D.A.; Jensen, C.D.; Marks, A.R.; Zhao, W.K.; Zebrowski, A.M.; Quinn, V.P.; Browne, L.W.; Taylor, W.R.; Ahlquist, D.A.; et al. Genetic Biomarker Prevalence is Similar in Fecal Immunochemical Test Positive and Negative Colorectal Cancer Tissue. Dig. Dis. Sci. 2017, 62, 678–688. [Google Scholar] [CrossRef]
- Chang, P.Y.; Chen, J.S.; Chang, N.C.; Chang, S.C.; Wang, M.C.; Tsai, S.H.; Wen, Y.H.; Tsai, W.S.; Chan, E.C.; Lu, J.J. NRAS germline variant G138R and multiple rare somatic mutations on APC in colorectal cancer patients in Taiwan by next generation sequencing. Oncotarget 2016, 7, 37566–37580. [Google Scholar] [CrossRef]
- Chang, P.Y.; Chen, J.S.; Chang, S.C.; Wang, M.C.; Chang, N.C.; Wen, Y.H.; Tsai, W.S.; Liu, W.H.; Liu, H.L.; Lu, J.J. Acquired somatic TP53 or PIK3CA mutations are potential predictors of when polyps evolve into colorectal cancer. Oncotarget 2017, 8, 72352–72362. [Google Scholar] [CrossRef]
- Lee, Y.C.; Li-Sheng, C.S.; Ming-Fang, Y.A.; Yueh-Hsia, C.S.; Ching-Yuan, F.J.; Chuang, S.L.; Chiang, T.H.; Chou, C.K.; Chiu, H.M.; Wu, M.S.; et al. Association Between Colorectal Cancer Mortality and Gradient Fecal Hemoglobin Concentration in Colonoscopy Noncompliers. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Kriegsmann, M.; Arens, N.; Endris, V.; Weichert, W.; Kriegsmann, J. Detection of KRAS, NRAS and BRAF by mass spectrometry—A sensitive, reliable, fast and cost-effective technique. Diagn. Pathol. 2015, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Changela, K.; Basi, P.; Mathur, S.; Reddy, S.; Momeni, M.; Krishnaiah, M.; Anand, S. Prescreening with FOBT Improves Yield and Is Cost-Effective in Colorectal Screening in the Elderly. ISRN Gastroenterol. 2014, 2014, 179291. [Google Scholar] [CrossRef]
- Doolittle, B.R.; Emanuel, J.; Tuttle, C.; Costa, J. Detection of the mutated K-Ras biomarker in colorectal carcinoma. Exp. Mol. Pathol. 2001, 70, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Youssef, O.L.; Sarhadi, V.; Ehsan, H.; Bohling, T.; Carpelan-Holmstrom, M.; Koskensalo, S.; Puolakkainen, P.; Kokkola, A.; Knuutila, S. Gene mutations in stool from gastric and colorectal neoplasia patients by next-generation sequencing. World J. Gastroenterol. 2017, 23, 8291–8299. [Google Scholar] [CrossRef]
- Miller, S.; Steele, S. Novel molecular screening approaches in colorectal cancer. J. Surg. Oncol. 2012, 105, 459–467. [Google Scholar] [CrossRef]
- Kogita, A.; Yoshioka, Y.; Sakai, K.; Togashi, Y.; Sogabe, S.; Nakai, T.; Okuno, K.; Nishio, K. Inter- and intra-tumor profiling of multi-regional colon cancer and metastasis. Biochem. Biophys. Res. Commun. 2015, 458, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Sievers, C.K.; Zou, L.S.; Pickhardt, P.J.; Matkowskyj, K.A.; Albrecht, D.M.; Clipson, L.; Bacher, J.; Pooler, B.D.; Moawad, F.J.; Cash, B.D.; et al. Subclonal diversity arises early even in small colorectal tumours and contributes to differential growth fates. Gut 2017, 66, 2132–2140. [Google Scholar] [CrossRef]
- Itzkowitz, S.; Brand, R.; Jandorf, L.; Durkee, K.; Millholland, J.; Rabeneck, L.; Schroy, P.C.; Sontag, S.; Johnson, D.; Markowitz, S.; et al. A simplified, noninvasive stool DNA test for colorectal cancer detection. Am. J. Gastroenterol. 2008, 103, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Milbury, C.A.; Li, J.; Makrigiorgos, G.M. PCR-based methods for the enrichment of minority alleles and mutations. Clin. Chem. 2009, 55, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Ahlquist, D.A. Next-generation stool DNA testing: Expanding the scope. Gastroenterology 2009, 136, 2068–2073. [Google Scholar] [CrossRef]
- Shin, S.J.; Chun, S.M.; Kim, T.I.; Kim, Y.J.; Choi, H.J.; Jang, S.J.; Kim, J. Feasibility of multiplexed gene mutation detection in plasma samples of colorectal cancer patients by mass spectrometric genotyping. PLoS ONE 2017, 12, e0176340. [Google Scholar] [CrossRef]
- Ahlquist, D.A.; Zou, H.; Domanico, M.; Mahoney, D.W.; Yab, T.C.; Taylor, W.R.; Butz, M.L.; Thibodeau, S.N.; Rabeneck, L.; Paszat, L.F.; et al. Next-generation stool DNA test accurately detects colorectal cancer and large adenomas. Gastroenterology 2012, 142, 248–256. [Google Scholar] [CrossRef]
- Catalogue of Somatic Mutations in Cancer. Available online: http://cancer.sanger.ac.uk/cosmic/ (accessed on 19 March 2019).
- Sakai, K.; Horiike, A.; Irwin, D.L.; Kudo, K.; Fujita, Y.; Tanimoto, A.; Sakatani, T.; Saito, R.; Kaburaki, K.; Yanagitani, N.; et al. Detection of epidermal growth factor receptor T790M mutation in plasma DNA from patients refractory to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci. 2013, 104, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Croke, L. Colorectal Cancer Screening: ACS Updates Guideline for Adults with Average Risk. Am. Fam. Phys. 2019, 99, 129–130. [Google Scholar]
- Ahmed, F.E.; Amed, N.C.; Vos, P.W.; Bonnerup, C.; Atkins, J.N.; Casey, M.; Nuovo, G.J.; Naziri, W.; Wiley, J.E.; Allison, R.R. Diagnostic microRNA markers to screen for sporadic human colon cancer in blood. Cancer Genom. Proteomics 2012, 9, 179–192. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | CDS Mutation | AA Mutation | CRC | Polyps | Mutation Frequency in COSMIC | Validated Sample ID (LOD%) |
|---|---|---|---|---|---|---|
| APC | c.2626C > T | p.R876* | 3 | 2 | 2% | CRC-1445 (1.65%) |
| c.3340C > T | p.R1114* | 2 | 1 | 1% | LoVo cell line (1.4%) | |
| c.3871C > T | p.Q1291* | 0 | 2 | <1% | Polyp-44111 (3.64%) | |
| c.3916G > T | p.E1306* | 0 | 2 | 1% | Polyp-36137 (2.8%) | |
| c.4348C > T | p.R1450* | 0 | 4 | 5% | Polyp-13849 (26%) | |
| c.4661dupA | p.E1554fs | 2 | 0 | <1% | CRC-1480 (14.9%) | |
| KRAS | c.34G > T/A | p.G12C/S | 1 | 2 | 77% | CRC-1773 (2.4%) |
| c.35G > T/A | p.G12V/D | 12 | 8 | CRC-1897(1.25%) | ||
| c.38G > A | p.G13D | 5 | 4 | 20% | LoVo cell line (1.4%) | |
| c.183A > C | p.Q61H | 1 | 2 | <1% | CRC-1897(1.75%) | |
| c.436G > A | p.A146T | 2 | 1 | <1% | CRC-1425(2.54%) | |
| TP53 | c.524G > A | p.R175H | 4 | 0 | 11% | CRC-1445(2.7%) |
| c.637C > T | p.R213* | 3 | 0 | 2% | CRC-1738(4.87%) | |
| PIK3CA | c.263G > A | p.R88Q | 2 | 0 | 1% | CRC-1773(2.58%) |
| c.1035T > A | p.N345K | 2 | 0 | <1% | CRC-1723(2.57%) | |
| c.1633G > A | p.E545K | 4 | 0 | 31% | CRC-1480(3.04%) | |
| c.3073A > G/C | p.T1025A/P | 2 | 0 | <1% | CRC-1801(1.99%) | |
| SMAD4 | c.1082G > A | p.R361H | 2 | 2 | 10% | CRC-1425(1.71%) |
| Features | Cohort 1 | Cohort 2 | ||||
|---|---|---|---|---|---|---|
| Normal | Polyps | CRC | Normal | Polyps | CRC | |
| Sample no | 9 | 13 | 21 | 20 | 27 | 54 |
| Gender (M/F) | 5/4 | 9/4 | 13/8 | 8/12 | 20/7 | 31/23 |
| Age (mean ± SD) | 64 ± 8 | 59 ± 7.6 | 68 ± 11.6 | 56.9 ± 8.9 | 65.3 ± 6.9 | 61.9 ± 11.1 |
| FIT positive rate (%) | 100% | 46% | 52% | 100% | 100% | 100% |
| Lesion location | ||||||
| Rectum | 1 | 8 | 7 | 11 | ||
| Left | 7 | 9 | 16 | 21 | ||
| Right | 5 | 4 | 4 | 22 | ||
| Lesion stage | ||||||
| Benign polyps | 3 | 19 | ||||
| Advanced polyps | 10 | 8 | ||||
| Early stage of CRC (stage I-II) | 11 | 27 | ||||
| Late stage of CRC (stage III-IV) | 10 | 27 | ||||
| Lesion size (max diameter) | ||||||
| <1 cm | 6 | 0 | 22 | 0 | ||
| 1–3 cm | 7 | 5 | 5 | 12 | ||
| >3 cm | 0 | 16 | 0 | 42 | ||
| Histological Classification | Sample ID | Sample Features | Expected Mutations by Stool Panel | Measured Mutations by Stool Panel | Concordance (Y/N) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stage | Location | Size-W (cm) | FIT | APC | KRAS | TP53 | PIK3CA | SMAD4 | APC | KRAS | TP53 | PIK3CA | SMAD4 | |||
| Adenoma | 91739 | Tubular | Left | 0.7 | + | Y | ||||||||||
| 45248 | Tubular | Left | 1.2 | − | p.R1114* | N | ||||||||||
| 88633 | Tubular | Right | 0.1 | + | p.E1306* | N | ||||||||||
| 30523 2 | Tubular | Right | 0.2 | + | Y | |||||||||||
| 17766 3 | Tubulovillous | Left | 0.9 | + | Y | |||||||||||
| 90511 | Tubulovillous | Left | 1.1 | − | p.Q61H | N | ||||||||||
| 18039 | Tubulovillous | Left | 2.2 | + | p.R876* | N | ||||||||||
| 44111 3 | Tubulovillous | Right | 1.4 | − | p.Q1291* | p.G12V | N | |||||||||
| 28808 | Tubulovillous | Right | 1.5 | − | p.G12D | N | ||||||||||
| 29563 | Villous | Rectum | 2.2 | − | p.R876* | p.G13D | N | |||||||||
| 60570 | Villous | Left | 1.7 | − | p.R1450* | p.G12S | N | |||||||||
| 34335 | Villous | Left | 0.1 | − | p.G12D/ p.G13D | N | ||||||||||
| 13849 | Villous | Right | 0.2 | + | p.R1450* | p.G12V | N | |||||||||
| Early stage of CRC | 1325 | II | Rectum | 3.5 | + | p.R213* | N | |||||||||
| 1387 | II | Rectum | 3.5 | − | p.G12V | p.E545K | p.E545K/ p.T1025A | Y | ||||||||
| 1736 | II | Rectum | 3.8 | + | p.G12V | p.G12V | Y | |||||||||
| 1454 | I | Left | 2 | − | p.R1114* | p.R1114* | Y | |||||||||
| 1317 | II | Left | 2.3 | + | Y | |||||||||||
| 1455 | I | Left | 3.1 | − | Y | |||||||||||
| 1465 | II | Left | 3.5 | + | p.G12V | p.G12V | Y | |||||||||
| 1371 | I | Left | 4 | + | p.R175H | N | ||||||||||
| 1423 | II | Left | 4.1 | − | p.G12D | N | ||||||||||
| 1801 | II | Right | 2.8 | + | p.R876* | p.G12V | p.T1025A | p.T1025A | Y | |||||||
| 1453 | II | Right | 8 | − | p.G13D | p.E545K | N | |||||||||
| Late stage of CRC | 1897 | III | Rectum | 2.7 | − | p.Q61H/ p.G12D | N | |||||||||
| 1425 | III | Rectum | 4 | + | p.A146T | p.R361H | N | |||||||||
| 1657 | III | Rectum | 4.2 | + | p.G12V | N | ||||||||||
| 1539 | IV | Rectum | 4.3 | − | Y | |||||||||||
| 1738 | III | Rectum | 4.8 | + | p.R213* | p.R213* | Y | |||||||||
| 1840 | III | Left | 3.4 | + | p.G12V | p.R175H | p.G12V | p.R175H | Y | |||||||
| 1480 | III | Left | 4.6 | − | p.E1554fs | p.G12D | p.E545K | p.E545K | Y | |||||||
| 1773 | III | Left | 5 | + | p.G12S | p.R88Q | p.G12S | Y | ||||||||
| 1445 | III | Right | 2.3 | − | p.R876* | p.R175H | p.R876* | Y | ||||||||
| 1723 | III | Right | 3.9 | − | p.R876* | p.N345K | N | |||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, P.-Y.; Chen, C.-C.; Chiang, J.-M.; Chang, S.-C.; Wang, M.-C.; Chen, J.-S.; Tsai, W.-S.; You, J.F.; Lu, J.-J. A Simple and Highly Specific MassARRAY-Based Stool DNA Assay to Prioritize Follow-up Decisions in Fecal Immunochemical Test-Positive Individuals. Cancers 2019, 11, 423. https://doi.org/10.3390/cancers11030423
Chang P-Y, Chen C-C, Chiang J-M, Chang S-C, Wang M-C, Chen J-S, Tsai W-S, You JF, Lu J-J. A Simple and Highly Specific MassARRAY-Based Stool DNA Assay to Prioritize Follow-up Decisions in Fecal Immunochemical Test-Positive Individuals. Cancers. 2019; 11(3):423. https://doi.org/10.3390/cancers11030423
Chicago/Turabian StyleChang, Pi-Yueh, Chia-Chun Chen, Jy-Ming Chiang, Shih-Cheng Chang, Mei-Chia Wang, Jinn-Shiun Chen, Wen-Sy Tsai, Jeng Fu You, and Jang-Jih Lu. 2019. "A Simple and Highly Specific MassARRAY-Based Stool DNA Assay to Prioritize Follow-up Decisions in Fecal Immunochemical Test-Positive Individuals" Cancers 11, no. 3: 423. https://doi.org/10.3390/cancers11030423
APA StyleChang, P.-Y., Chen, C.-C., Chiang, J.-M., Chang, S.-C., Wang, M.-C., Chen, J.-S., Tsai, W.-S., You, J. F., & Lu, J.-J. (2019). A Simple and Highly Specific MassARRAY-Based Stool DNA Assay to Prioritize Follow-up Decisions in Fecal Immunochemical Test-Positive Individuals. Cancers, 11(3), 423. https://doi.org/10.3390/cancers11030423