Mutated Rnf43 Aggravates Helicobacter Pylori-Induced Gastric Pathology


Abstract
1. Introduction
2. Results
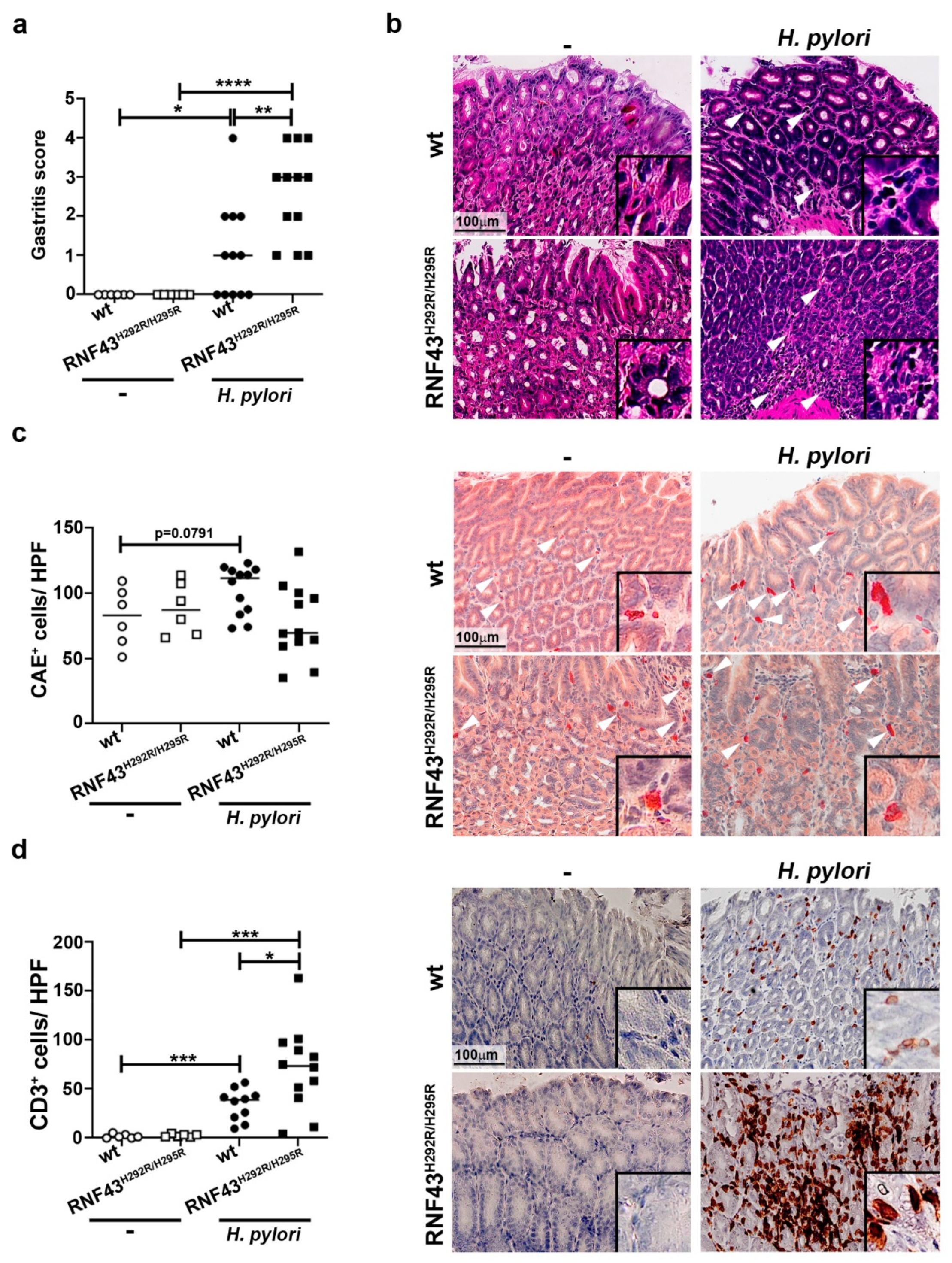
2.1. RNF43H292R/H295R Mice Show Enhanced H. pylori-Induced Gastritis Inflammation
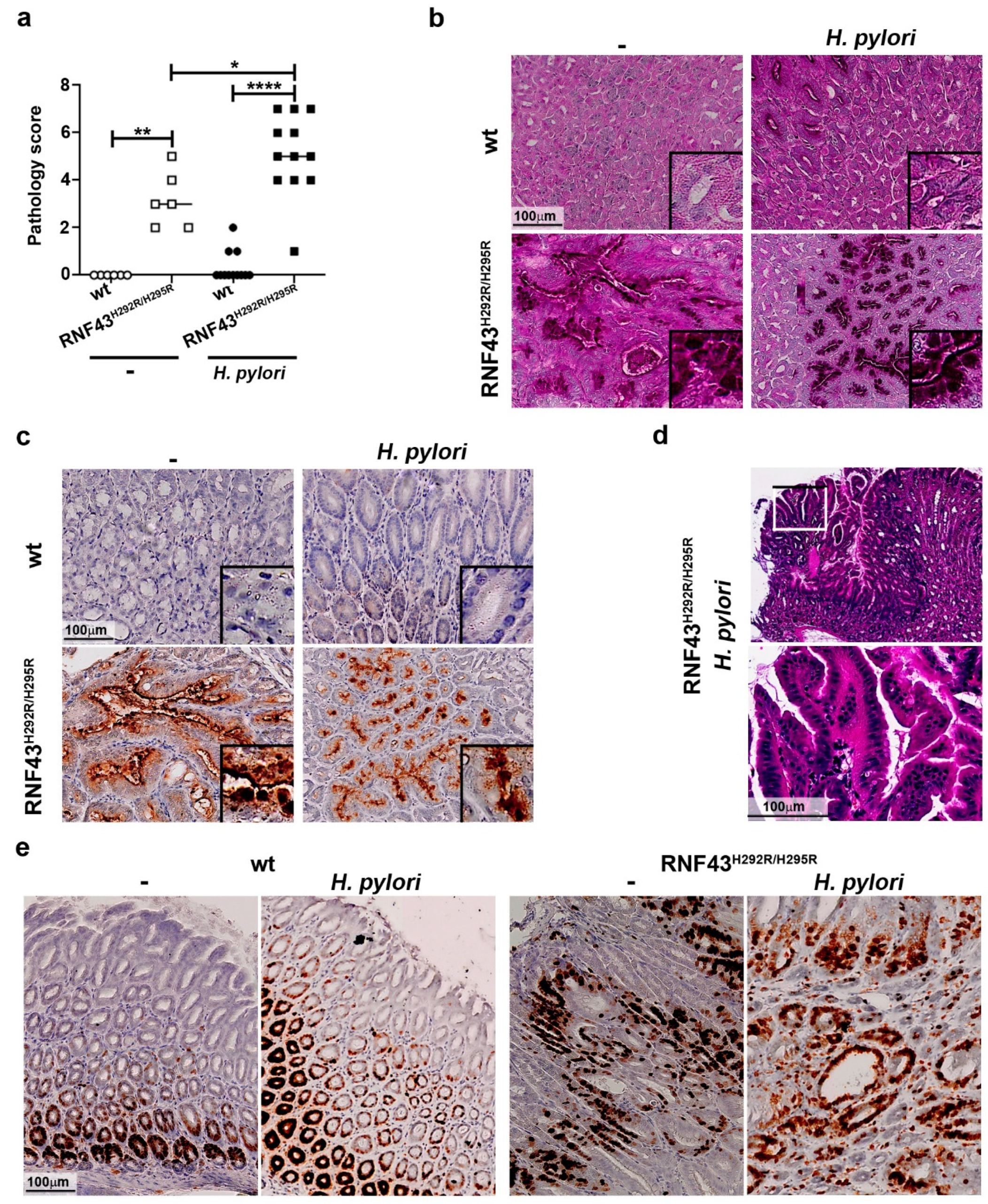
2.2. H. pylori Increases Gastric Pathology of RNF43H292R/H295R Mice
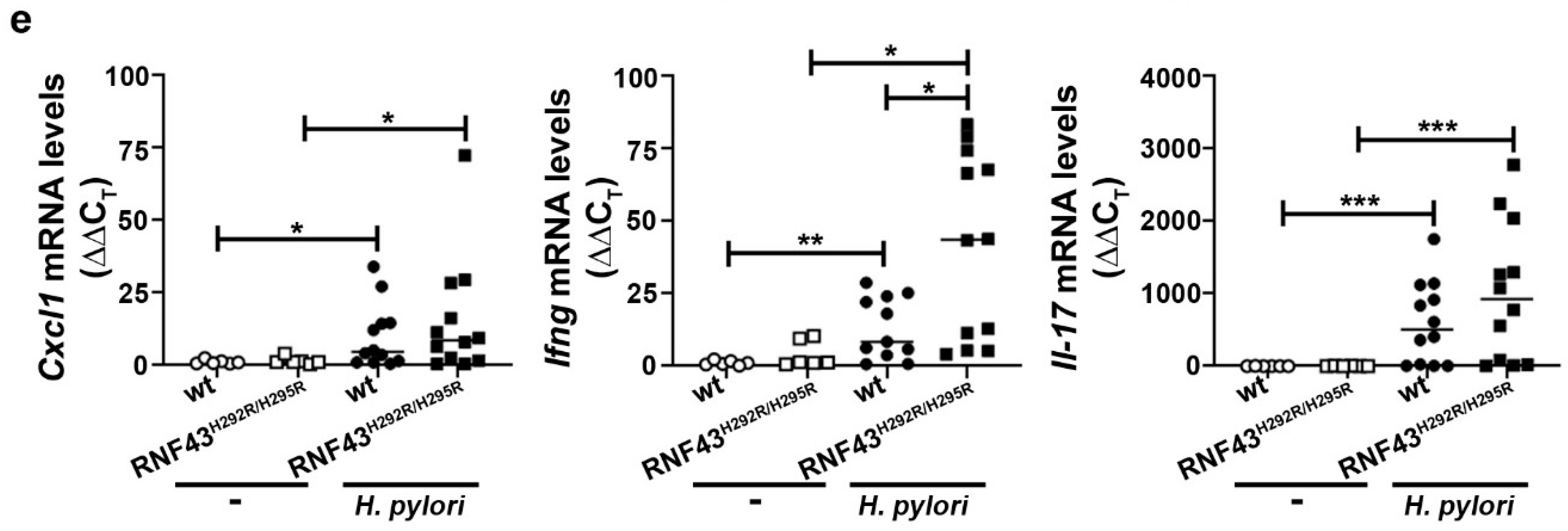
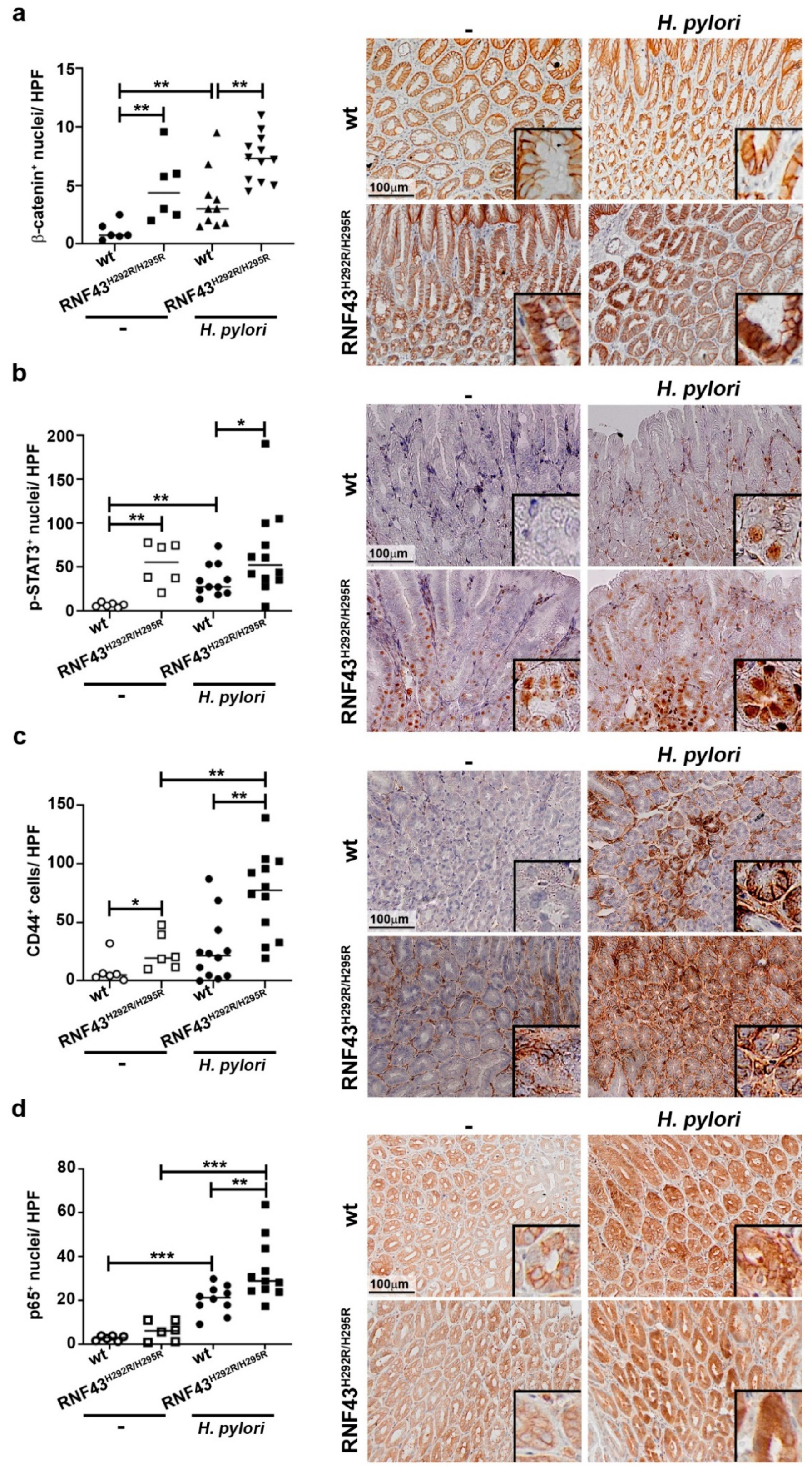
2.3. RNF43 Transactivating Mutations Favor the Expression of Genes Related to Gastric Carcinogenesis
3. Discussion
4. Materials and Methods
4.1. Helicobacter Pylori Infection
4.2. Quantitative PCR
4.3. Immunohistochemistry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aichler, M.; Luber, B.; Lordick, F.; Walch, A. Proteomic and metabolic prediction of response to therapy in gastric cancer. World J. Gastroenterol. 2014, 20, 13648–13657. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nature Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Maruvka, Y.E.; Mouw, K.W.; Karlic, R.; Parasuraman, P.; Kamburov, A.; Polak, P.; Haradhvala, N.J.; Hess, J.M.; Rheinbay, E.; Brody, Y.; et al. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nature Biotechnol. 2017, 35, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Qin, H.Z.; Xi, H.Q.; Wei, B.; Xia, S.Y.; Chen, L. RNF43 inhibits cancer cell proliferation and could be a potential prognostic factor for human gastric carcinoma. Cell. Physiol. Biochem. 2015, 36, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Min, B.H.; Hwang, J.; Kim, N.K.; Park, G.; Kang, S.Y.; Ahn, S.; Ahn, S.; Ha, S.Y.; Lee, Y.K.; Kushima, R.; et al. Dysregulated WNT signalling and recurrent mutations of the tumour suppressor RNF43 in early gastric carcinogenesis. J. Pathol. 2016, 10. [Google Scholar] [CrossRef]
- Gao, Y.; Cai, A.; Xi, H.; Li, J.; Xu, W.; Zhang, Y.; Zhang, K.; Cui, J.; Wu, X.; Wei, B.; et al. Ring finger protein 43 associates with gastric cancer progression and attenuates the stemness of gastric cancer stem-like cells via the Wnt-beta/catenin signaling pathway. Stem Cell Res. Ther. 2017, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Neumeyer, V.; Grandl, M.; Dietl, A.; Brutau-Abia, A.; Allgauer, M.; Kalali, B.; Zhang, Y.; Pan, K.F.; Steiger, K.; Vieth, M.; et al. Loss of endogenous RNF43 function enhances proliferation and tumour growth of intestinal and gastric cells. Carcinogenesis 2018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Lu, B.; Hsieh, M.H.; Bagdasarian, L.; Meyer, R.; et al. Inactivating mutations of RNF43 confer WNT dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12649–12654. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of WNT receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef]
- Loregger, A.; Grandl, M.; Mejias-Luque, R.; Allgauer, M.; Degenhart, K.; Haselmann, V.; Oikonomou, C.; Hatzis, P.; Janssen, K.P.; Nitsche, U.; et al. The E3 ligase RNF43 inhibits WNT signaling downstream of mutated beta-catenin by sequestering TCF4 to the nuclear membrane. Sci. Signal. 2015, 8, ra90. [Google Scholar] [CrossRef]
- Anonymous. Infection with Helicobacter pylori. IARC Monogr. Eval. Carcinog. Risks. Hum. 1994, 61, 177–240. [Google Scholar]
- Mejias-Luque, R.; Gerhard, M. Immune evasion strategies and persistence of Helicobacter pylori. Curr. Top. Microbiol. Immunol. 2017, 400, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Kandulski, A.; Malfertheiner, P.; Wex, T. Role of regulatory T-cells in H. pylori-induced gastritis and gastric cancer. Anticancer Res. 2010, 30, 1093–1103. [Google Scholar] [PubMed]
- Capitani, N.; Codolo, G.; Vallese, F.; Minervini, G.; Grassi, A.; Cianchi, F.; Troilo, A.; Fischer, W.; Zanotti, G.; Baldari, C.T.; et al. The lipoprotein HP1454 of Helicobacter pylori regulates T-cell response by shaping T-cell receptor signalling. Cell. Microbiol. 2019, e13006. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society award lecture on cancer epidemiology and prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Lamb, A.; Chen, L.F. Role of the Helicobacter pylori-induced inflammatory response in the development of gastric cancer. J. Cell. Biochem. 2013, 114, 491–497. [Google Scholar] [CrossRef]
- Mejias-Luque, R.; Zoller, J.; Anderl, F.; Loew-Gil, E.; Vieth, M.; Adler, T.; Engler, D.B.; Urban, S.; Browning, J.L.; Muller, A.; et al. Lymphotoxin beta receptor signalling executes Helicobacter pylori-driven gastric inflammation in a T4SS-dependent manner. Gut 2017, 66, 1369–1381. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Asim, M.; Piazuelo, M.B.; Yan, F.; Barry, D.P.; Sierra, J.C.; Delgado, A.G.; Hill, S.; Casero, R.A., Jr.; Bravo, L.E.; et al. Activation of EGFR and ERBB2 by Helicobacter pylori results in survival of gastric epithelial cells with DNA damage. Gastroenterology 2014, 146, 1739–1751. [Google Scholar] [CrossRef]
- Sierra, J.C.; Asim, M.; Verriere, T.G.; Piazuelo, M.B.; Suarez, G.; Romero-Gallo, J.; Delgado, A.G.; Wroblewski, L.E.; Barry, D.P.; Peek, R.M., Jr.; et al. Epidermal growth factor receptor inhibition downregulates Helicobacter pylori-induced epithelial inflammatory responses, DNA damage and gastric carcinogenesis. Gut 2017. [Google Scholar] [CrossRef]
- Wen, J.; Wang, Y.; Gao, C.; Zhang, G.; You, Q.; Zhang, W.; Zhang, Z.; Wang, S.; Peng, G.; Shen, L. Helicobacter pylori infection promotes Aquaporin 3 expression via the ROS-HIF-1alpha-AQP3-ROS loop in stomach mucosa: A potential novel mechanism for cancer pathogenesis. Oncogene 2018, 37, 3549–3561. [Google Scholar] [CrossRef] [PubMed]
- Sigal, M.; Rothenberg, M.E.; Logan, C.Y.; Lee, J.Y.; Honaker, R.W.; Cooper, R.L.; Passarelli, B.; Camorlinga, M.; Bouley, D.M.; Alvarez, G.; et al. Helicobacter pylori activates and expands Lgr5(+) stem cells through direct colonization of the gastric glands. Gastroenterology 2015, 148, 1392–1404.e21. [Google Scholar] [CrossRef]
- Khurana, S.S.; Riehl, T.E.; Moore, B.D.; Fassan, M.; Rugge, M.; Romero-Gallo, J.; Noto, J.; Peek, R.M., Jr.; Stenson, W.F.; Mills, J.C. The hyaluronic acid receptor CD44 coordinates normal and metaplastic gastric epithelial progenitor cell proliferation. J. Biological Chem. 2013, 288, 16085–16097. [Google Scholar] [CrossRef]
- Dixon, M.F.; Genta, R.M.; Yardley, J.H.; Correa, P. Classification and grading of gastritis. The updated Sydney system. International workshop on the histopathology of gastritis, Houston 1994. Am. J. Surg. Pathol. 1996, 20, 1161–1181. [Google Scholar] [CrossRef]
- Carrasco-Garcia, E.; Santos, J.C.; Garcia, I.; Brianti, M.; Garcia-Puga, M.; Pedrazzoli, J., Jr.; Matheu, A.; Ribeiro, M.L. Paradoxical role of SOX2 in gastric cancer. Am. J. Cancer Res. 2016, 6, 701–713. [Google Scholar]
- Jenkins, B.J.; Grail, D.; Nheu, T.; Najdovska, M.; Wang, B.; Waring, P.; Inglese, M.; McLoughlin, R.M.; Jones, S.A.; Topley, N.; et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat. Med. 2005, 11, 845–852. [Google Scholar] [CrossRef]
- Bertaux-Skeirik, N.; Feng, R.; Schumacher, M.A.; Li, J.; Mahe, M.M.; Engevik, A.C.; Javier, J.E.; Peek, R.M., Jr.; Ottemann, K.; Orian-Rousseau, V.; et al. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog. 2015, 11, e1004663. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Chiurillo, M.A. Role of the WNT/beta-catenin pathway in gastric cancer: An in-depth literature review. World J. Exp. Med. 2015, 5, 84–102. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.L.; Xiang, D.; Shigdar, S.; Macdonald, J.; Li, Y.; Wang, T.; Pu, C.; Wang, Z.; Qiao, L.; Duan, W. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014, 344, 180–187. [Google Scholar] [CrossRef]
- Brungs, D.; Aghmesheh, M.; Vine, K.L.; Becker, T.M.; Carolan, M.G.; Ranson, M. Gastric cancer stem cells: Evidence, potential markers, and clinical implications. J. Gastroenterol. 2016, 51, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.; El-Rayes, B. Identifying and targeting cancer stem cells in the treatment of gastric cancer. Cancer 2017, 123, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, C.; He, F.; Cai, Y.; Yang, H. Identification of CD44+CD24+ gastric cancer stem cells. J. Cancer Res. Clin. Oncol. 2011, 137, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Garay, J.; Piazuelo, M.B.; Majumdar, S.; Li, L.; Trillo-Tinoco, J.; Del Valle, L.; Schneider, B.G.; Delgado, A.G.; Wilson, K.T.; Correa, P.; et al. The homing receptor CD44 is involved in the progression of precancerous gastric lesions in patients infected with Helicobacter pylori and in development of mucous metaplasia in mice. Cancer Lett. 2016, 371, 90–98. [Google Scholar] [CrossRef]
- Ishimoto, T.; Oshima, H.; Oshima, M.; Kai, K.; Torii, R.; Masuko, T.; Baba, H.; Saya, H.; Nagano, O. CD44+ slow-cycling tumor cell expansion is triggered by cooperative actions of WNT and prostaglandin E2 in gastric tumorigenesis. Cancer Sci. 2010, 101, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Fan, S.; Ma, W.; Fan, P.; Wang, B.; Zhang, J.; Wang, H.; Tang, B.; Zhang, Q.; Yu, X.; et al. Roles of WNT/beta-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 2014, 5, e1039. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Israel, D.A.; Washington, M.K.; Krishna, U.; Fox, J.G.; Rogers, A.B.; Neish, A.S.; Collier-Hyams, L.; Perez-Perez, G.I.; Hatakeyama, M.; et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2005, 102, 10646–10651. [Google Scholar] [CrossRef] [PubMed]
- Nagy, T.A.; Wroblewski, L.E.; Wang, D.; Piazuelo, M.B.; Delgado, A.; Romero-Gallo, J.; Noto, J.; Israel, D.A.; Ogden, S.R.; Correa, P.; et al. beta-Catenin and p120 mediate PPARdelta-dependent proliferation induced by Helicobacter pylori in human and rodent epithelia. Gastroenterology 2011, 141, 553–564. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Piazuelo, M.B.; Chaturvedi, R.; Schumacher, M.; Aihara, E.; Feng, R.; Noto, J.M.; Delgado, A.; Israel, D.A.; Zavros, Y.; et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut 2015, 64, 720–730. [Google Scholar] [CrossRef]
- Smith, S.M.; Cai, L. Cell specific CD44 expression in breast cancer requires the interaction of AP-1 and NFkappaB with a novel cis-element. PLoS ONE 2012, 7, e50867. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Lyu, Y.L.; Cai, L. NF-kappaB affects proliferation and invasiveness of breast cancer cells by regulating CD44 expression. PLoS ONE 2014, 9, e106966. [Google Scholar] [CrossRef] [PubMed]
- Haria, D.; Trinh, B.Q.; Ko, S.Y.; Barengo, N.; Liu, J.; Naora, H. The homeoprotein DLX4 stimulates NF-kappaB activation and CD44-mediated tumor-mesothelial cell interactions in ovarian cancer. Am. J. Pathol. 2015, 185, 2298–2308. [Google Scholar] [CrossRef]
- Keates, S.; Hitti, Y.S.; Upton, M.; Kelly, C.P. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology 1997, 113, 1099–1109. [Google Scholar] [CrossRef]
- Shibata, W.; Takaishi, S.; Muthupalani, S.; Pritchard, D.M.; Whary, M.T.; Rogers, A.B.; Fox, J.G.; Betz, K.S.; Kaestner, K.H.; Karin, M.; et al. Conditional deletion of IkappaB-kinase-beta accelerates helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenterology 2010, 138, 1022–1034.e1. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, M.D.; Williams, J.M.; Duckworth, C.A.; O’Hara, A.; Hanedi, A.; Varro, A.; Caamano, J.H.; Pritchard, D.M. Signaling mediated by the NF-kappaB sub-units NF-kappaB1, NF-kappaB2 and c-Rel differentially regulate Helicobacter felis-induced gastric carcinogenesis in C57BL/6 mice. Oncogene 2013, 32, 5563–5573. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; O’Rourke, J.; Ungria, M.C.d.; Robertson, B.; Daskalopoulos, G.; Dixon, M.F. A standardized mouse model of Helicobacter pylori infection: Introducing the Sydney strain. Gastroenterology 1997, 112, 1386–1397. [Google Scholar] [CrossRef]
- Carlstedt, I.; Herrmann, A.; Hovenberg, H.; Lindell, G.; Nordman, H.; Wickstrom, C.; Davies, J.R. ‘Soluble’ and ‘insoluble’ mucins—Identification of distinct populations. Biochem. Soc. Trans. 1995, 23, 845–851. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward | Reverse |
|---|---|---|
| Cxcl10 | AAGTGCTGCCGTCATTTTCT | CCTATGGCCCTCATTCTCAC |
| Cxcl13 | ATATGTGTGAATCCTCGTGCCA | GGGAGTTGAAGACAGACTTTTGC |
| Gapdh | GCCTTCTCCATGGTGGTGAA | GCACAGTCAAGGCCGAGAAT |
| Ifng | TCAAGTGGCATAGATGTGGAAGAA | TGGCTCTGCAGGATTTTCATG |
| Il-17 | GCTCCAGAAGGCCCTCAGA | AGCTTTCCCTCCGCATTGA |
| Cxcl1 | TGCACCCAAACCGAAGTCAT | TTGTCAGAAGCCAGCGTTCAC |
| Sox2 | CATGGGCTCTGTGGTCAAGT | CGGGGAGGTACATGCTGATC |
| Target | Clone | Company |
|---|---|---|
| CD3 | SP7 | Thermo Fisher |
| CD44 | E7K2Y | Cell Signaling |
| MUC2 | LUM 2.3 | [48] |
| p-STAT3 | D3A7 | Cell Signaling |
| p65 | D14E12 | Cell Signaling |
| SOX2 | C70B1 | Cell Signaling |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumeyer, V.; Vieth, M.; Gerhard, M.; Mejías-Luque, R. Mutated Rnf43 Aggravates Helicobacter Pylori-Induced Gastric Pathology. Cancers 2019, 11, 372. https://doi.org/10.3390/cancers11030372
Neumeyer V, Vieth M, Gerhard M, Mejías-Luque R. Mutated Rnf43 Aggravates Helicobacter Pylori-Induced Gastric Pathology. Cancers. 2019; 11(3):372. https://doi.org/10.3390/cancers11030372
Chicago/Turabian StyleNeumeyer, Victoria, Michael Vieth, Markus Gerhard, and Raquel Mejías-Luque. 2019. "Mutated Rnf43 Aggravates Helicobacter Pylori-Induced Gastric Pathology" Cancers 11, no. 3: 372. https://doi.org/10.3390/cancers11030372
APA StyleNeumeyer, V., Vieth, M., Gerhard, M., & Mejías-Luque, R. (2019). Mutated Rnf43 Aggravates Helicobacter Pylori-Induced Gastric Pathology. Cancers, 11(3), 372. https://doi.org/10.3390/cancers11030372