1. Introduction
Lung cancer is one of the most malignant cancers with high incidence and mortality rates worldwide. Among the classified cancers, non-small cell lung cancer (NSCLC) accounts for 85% of lung cancer cases, and its incidence has continued to increase in the past decade. The prognosis of patients with NSCLC is poor, and over 50% of patients present with metastatic disease at diagnosis. Lung cancer is classified into two main types of resistance, primary and secondary. Platinum-based chemotherapy is recommended as the standard treatment for patients with malignant NSCLC. However, even with traditional radiochemical, chemotherapy or novel targeted therapy treatment, secondary resistance associated with various genetic mutations is a major problem accompanied by treatment failure and death [
1,
2].
Autophagy is an essential homeostatic process where cells break down their components for survival, differentiation, development, and homeostasis [
3]. While apoptosis is known as an active, programmed and very regulated form of cell death executed by caspases [
4], autophagy is considered a survival process but can be involved in mediating non-apoptotic cell death under certain conditions [
5,
6]. The main function of autophagy is to control cellular protein turnover, which is usually considered a constitutive mechanism that responds to several stress conditions [
7,
8]. The most important functions of autophagy are to prevent the accumulation of cellular debris that prohibits cytosolic and organelle homeostasis and to recycle cell components during nutrient starvation. In the latter case, autophagy acts more like a protective mechanism that guarantees cellular processes under limited or stressful environmental conditions [
9]. The degraded materials are further recycled for new biosynthetic and metabolic processes.
Furthermore, the regulation of autophagy and apoptosis is intimately connected; autophagy can inhibit apoptosis [
10], but long-term activation of autophagy can also lead to apoptotic cell death [
11]. Autophagy is divided into three main sequential steps, beginning with the initiation step by forming double-membrane vacuoles, called autophagosomes, capturing long-lived, misfolded or damaged proteins and aberrant organelles. Next is the maturation step, when cargo-containing vesicles fuse with lysosomes, leading to the final step of degradation and recycling of cellular constituents [
12]. These processes are regulated by different regulators; mammalian target of rapamycin (mTOR) and the Unc-51-like autophagy activating kinase (ULK1) complex initiate the formation of the phagophore and react with several autophagy-related genes (ATGs), followed by the maturation step of autophagosome sequestration and fusion with the lysosome by LC3 and LAMP2 activation and the final degradation step, with the formation of the autophagolysosome, which degrades the interior contents.
Various compounds inhibit autophagy progression at each step, and their mechanisms may differ. However, chloroquine (CQ), which is an anti-malarial drug approved for clinical use, was developed to inhibit autophagy and its related up-and downstream factors in many malignant tumors [
13,
14].
We previously reported the anticancer effect of C
2-ceramide and its additive effect upon combined treatment with paclitaxel [
15]. Treatment with C
2-ceramide alone induced cell apoptosis and arrested the cell cycle with AKT dephosphorylation [
16]. In addition, C
2-ceramide efficiently sensitized lung cancer cells to paclitaxel-induced senescence via a p21- and p16-independent pathway [
15]. An autophagy-dependent cell death pathway may also be involved in its anticancer effect, and its combination with another drug elevates its cytotoxic effect and reduces adverse effects; however, the mechanism has not yet been discovered.
In this study, we aimed to investigate the potential therapeutic effect of C2-ceramide combined with the autophagy inhibitor CQ on H460 and H1299 cells, which are representative of different types of NSCLC. Although we demonstrated that C2-ceramide is an apoptosis-inducing agent in lung cancer cells, it is important to reduce the needed dose without reducing its anticancer effects. Moreover, the underlying mechanism of this combined treatment on NSCLC cells is undefined and is worth further investigation.
3. Discussion
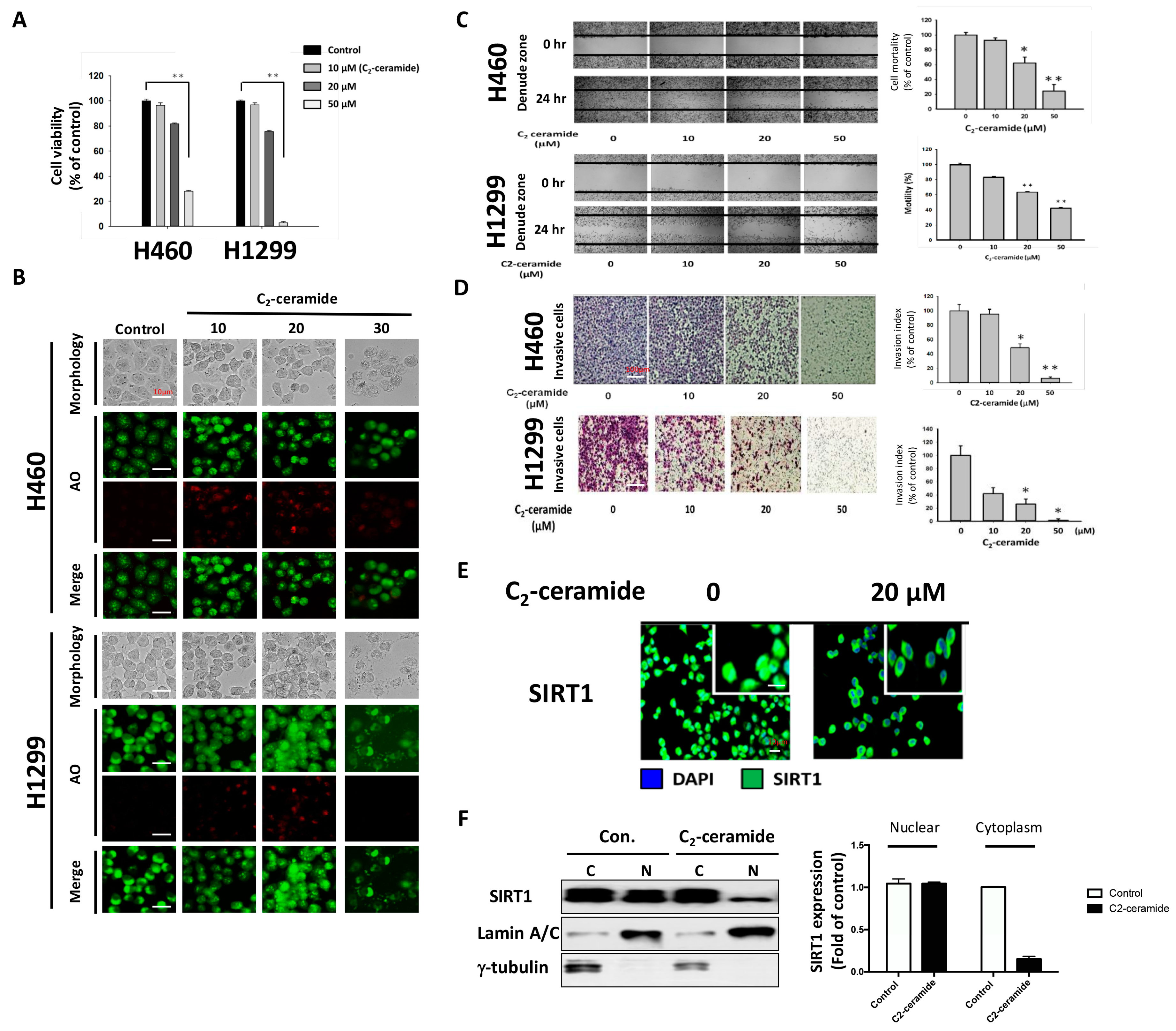
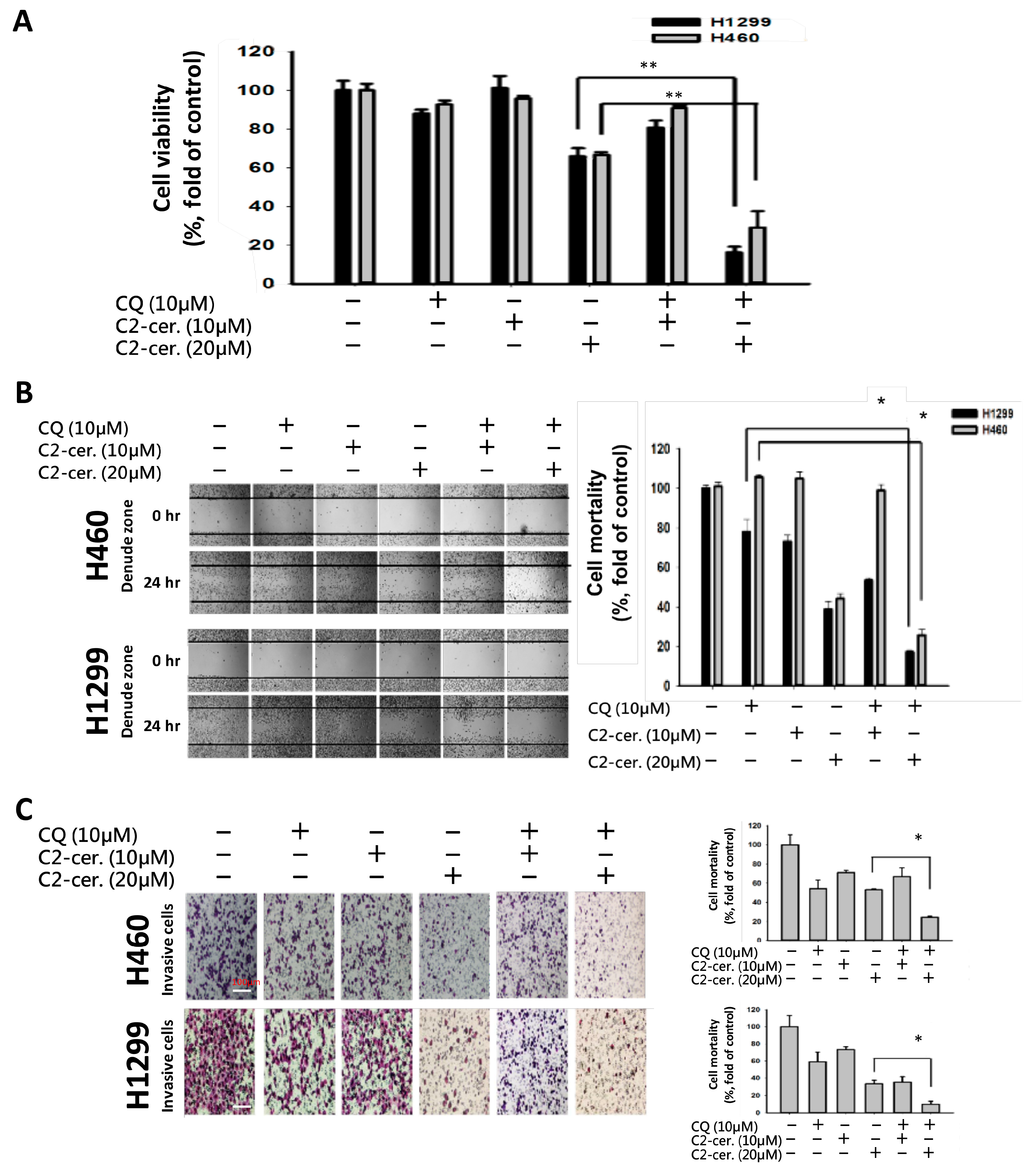
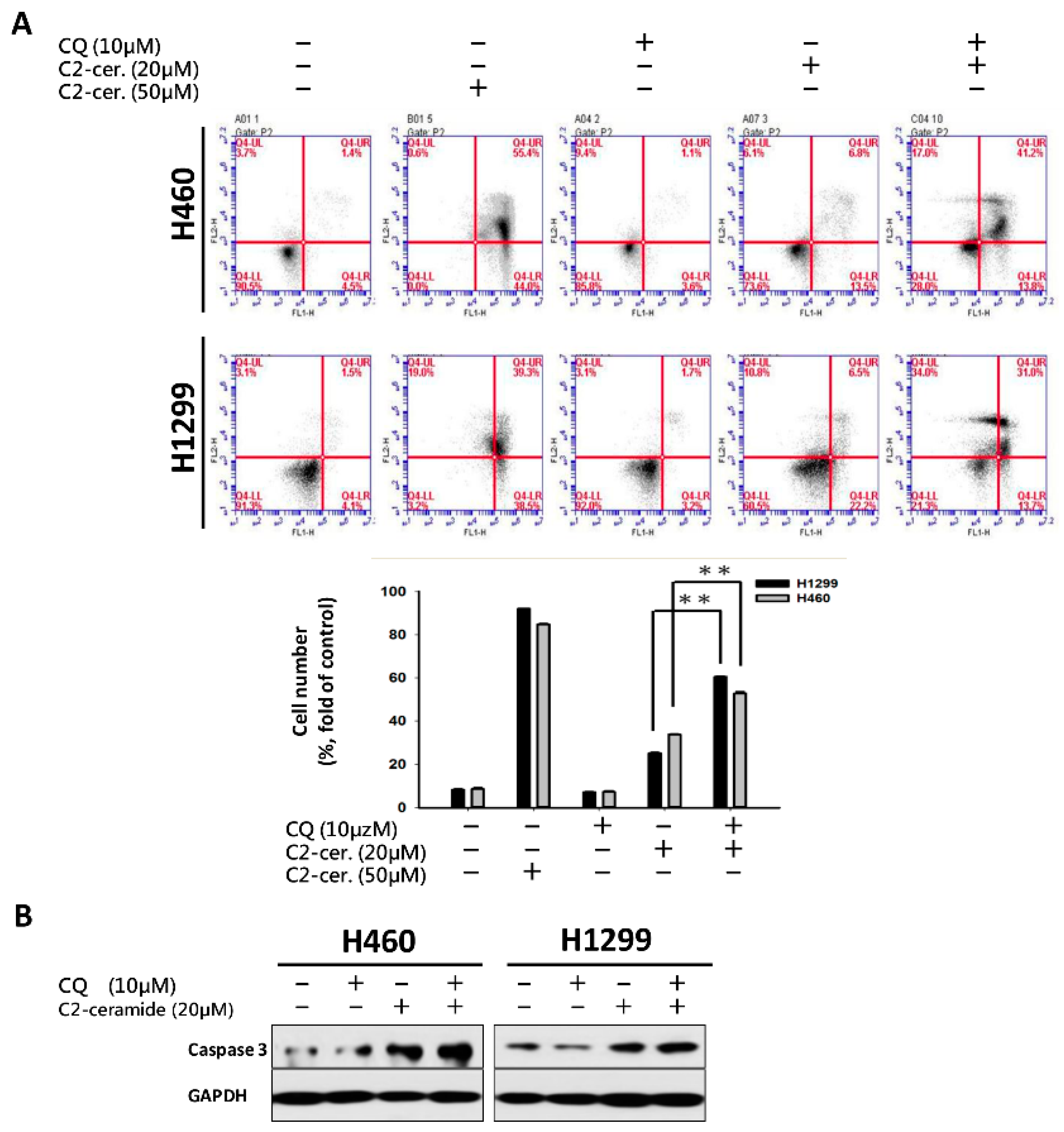
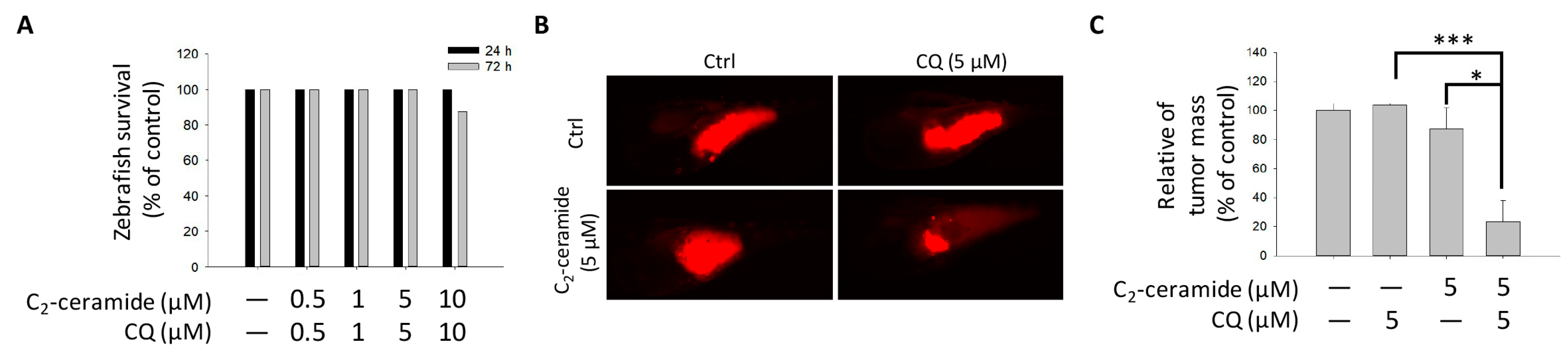
For the first time, we demonstrated the combinational effect of an autophagy inducer, C2-ceramide, and an autophagy inhibitor, CQ, in NSCLC cell lines. A single treatment with CQ can be expected to inhibit the autophagy process that blocks the autophagic apoptosis induced by C2-ceramide. However, cotreatment with a sublethal dose of C2-ceramide and CQ increased cytotoxicity and elevated autophagy signals in H460 and H1299 cells. The combined treatment enhanced the cytotoxicity of C2-ceramide by approximately 3.5- to 4.5-fold in the two cell lines, which increased cell death by 60% compared with single treatment. The enhanced cytotoxicity of combined treatment might arise via several mechanisms, including the following three: (1) reduced expression of the LC3 binding protein SIRT1, especially in the nucleus; (2) increased expression of SQSMT1 and the autophagy factor LC3 in the cytoplasm; and (3) inhibition of the Src tyrosine kinase pathway. The cytotoxicity of combination treatment significantly inhibited several cell behaviors, including cell proliferation, migration, and invasion, and induced severe apoptotic cell death. In addition, our in vivo analysis demonstrated that cotreatment with C2-ceramide and CQ enhanced a significant tumor-inhibition effect in the zebrafish xenograft model compared with single treatment groups, suggesting that the combined treatment was reliable for lung cancer treatment.
The anti-cancer effects of C
2-ceramide and its additive effects, when combined with clinical drugs, have been reported previously [
15]. In this study, the mechanism of autophagy and its utilization were the main aims to discover the novel anti-cancer drug in a different orientation. Sensitizing a cancer cell to a drug is always a critical issue in chemotherapy and chemoprevention, especially in the treatment of malignant NSCLC. C
2-Ceramide has proven cytotoxic effects against lung cancer. However, the IC
50 value was relatively high compared with clinical chemotherapy drugs, such as cisplatin and paclitaxel. It will be important to reduce the dose of C
2-ceramide in NSCLC treatment. Our findings suggest that although CQ is an autophagy inhibitor, the combined use of these two drugs at low doses can reach the same treatment efficacy of high dose C
2-ceramide in chemotherapy. In our study, the IC
50 of C
2-ceramide in H1299 and H460 was approximately 31 and 42 µM, respectively. In using a sublethal dose much lower than the IC
50 for combined usage, the additive effect of C
2-ceramide with a low concentration of CQ was apparent, enhancing cytotoxicity. In addition, the IC
50 of C
2-ceramide for H1299 and H460 was reduced to approximately 13 and 15 µM, respectively, under cotreatment with 10 µM CQ for 24 h. Most importantly, the safety of the combined treatment was further proved in normal lung cells. By combined treated Beas-2B and MRC-5 cells with indicated drugs, it showed low cytotoxicity toward normal lung cells below 40 µM of each drug combined use, with 85–95% cells remained survived in both normal lung cell lines (
Figure S1). The results also responded to the safety aspect of the findings in the zebrafish xenograft model. Combined these two drugs with sublethal dose exhibited great tumor-growth inhibition effect than along treatment, with extremely limited toxicity in zebrafish larvae, suggesting that the combined treatment strategy in lung cancer is relatively reliable by its enhanced cytotoxicity in tumor cells and safety to normal cells.
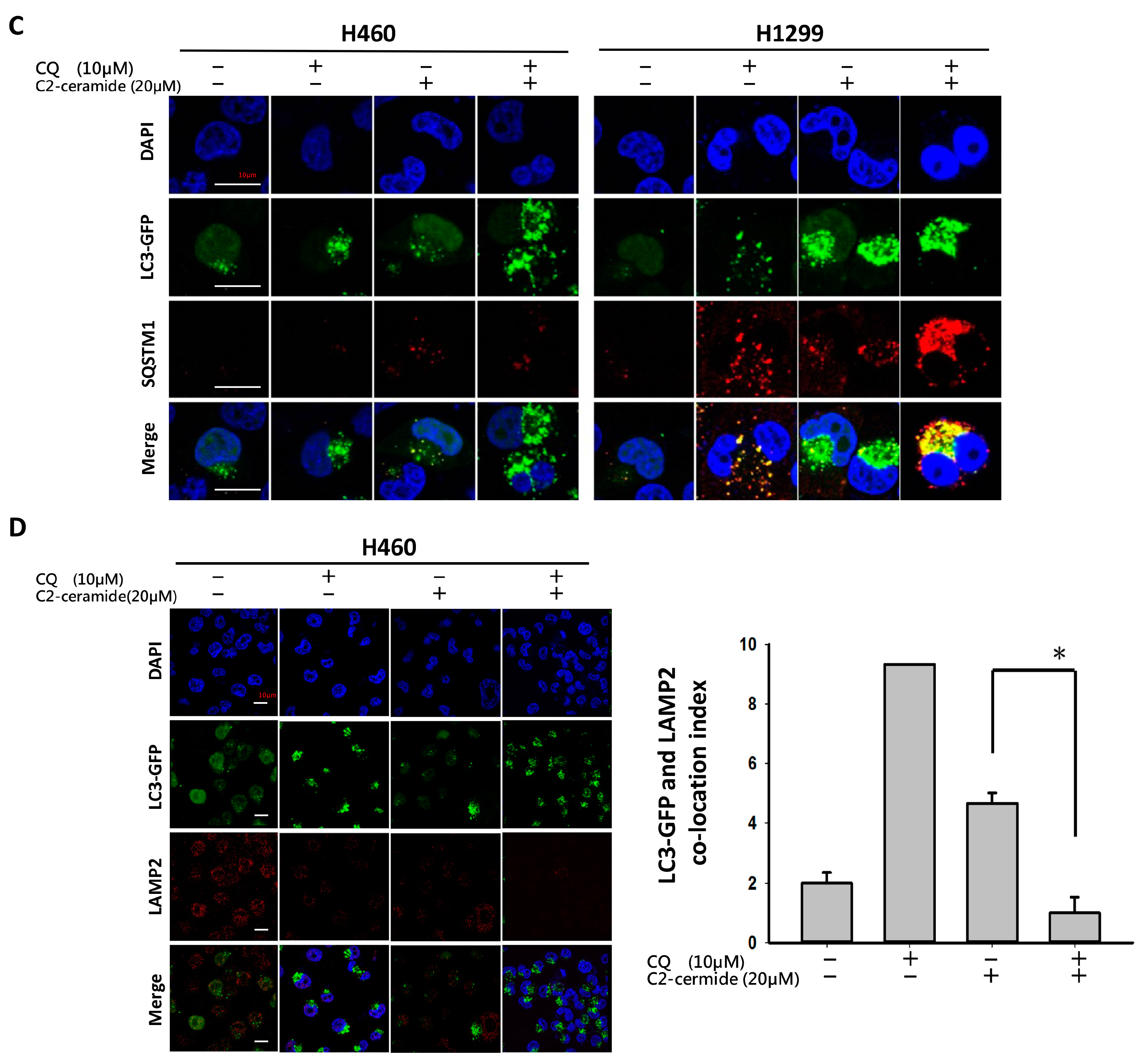
CQ inhibits lysosomal acidification and fusion with the autophagosome to prevent its degradation during late autophagy, thereby suppressing autophagy progression [
18]. CQ may have interfered with the normal autophagy process in our case, whereas C
2-ceramide continued to activate autophagy in NSCLC cells. The conflict between the inhibition and activation of autophagy occurred in the treated cells and was reflected in cell survival. The sublethal dose of the individual drug only slightly initiated apoptosis and autophagy; however, the additive effect induced advanced cell death and AO-dense autophagic cells. The increased cytotoxicity was also reflected in cell behavior; treatment not only induced a 60% increase in cell death relative to individual treatment but also caused a 20% to 30% enhancement in the suppression of cell migration and invasion. The enhancement was confirmed to increase cytotoxicity in vitro and in vivo, and the mechanisms involved were investigated.
Autophagy progresses in sequential stages governed by different regulators: (1) an initiation stage, (2) a maturation stage and (3) a degradation stage [
19]. Treatment with C
2-ceramide alone activated broad signal transduction, such as AKT dephosphorylation and deactivation of mTOR. mTOR acts as an important initiator of autophagy, which represses ULK1 complex formation to block the subsequent autophagy process. Our previous results suggested that ceramide inhibits AKT phosphorylation [
16]; such inhibition may deactivate its downstream target, mTOR, and initiate autophagy. Once the autophagic effect is initiated, the ULK1 complex and a group of ATGs are activated to promote autophagophore membrane formation and nucleation. Other important regulators, LC3 I/II, which were both activated following cotreatment with C
2-ceramide and CQ, affect the maturation of the autophagosome. LC3, especially subunit II, was over-expressed following cotreatment with C
2-ceramide and CQ relative to single treatment. These results indicate another feedback mechanism or an additional effect in regulating LC3 or ATG expression. The effect of CQ on the inhibition of lysosomes fusing to autophagosomes demonstrates that autophagosome degradation was prevented in the last stage of autophagy. The accumulation of autophagy initiation and the non-degraded autophagosome create enormous stress that may ultimately collapse the cellular process.
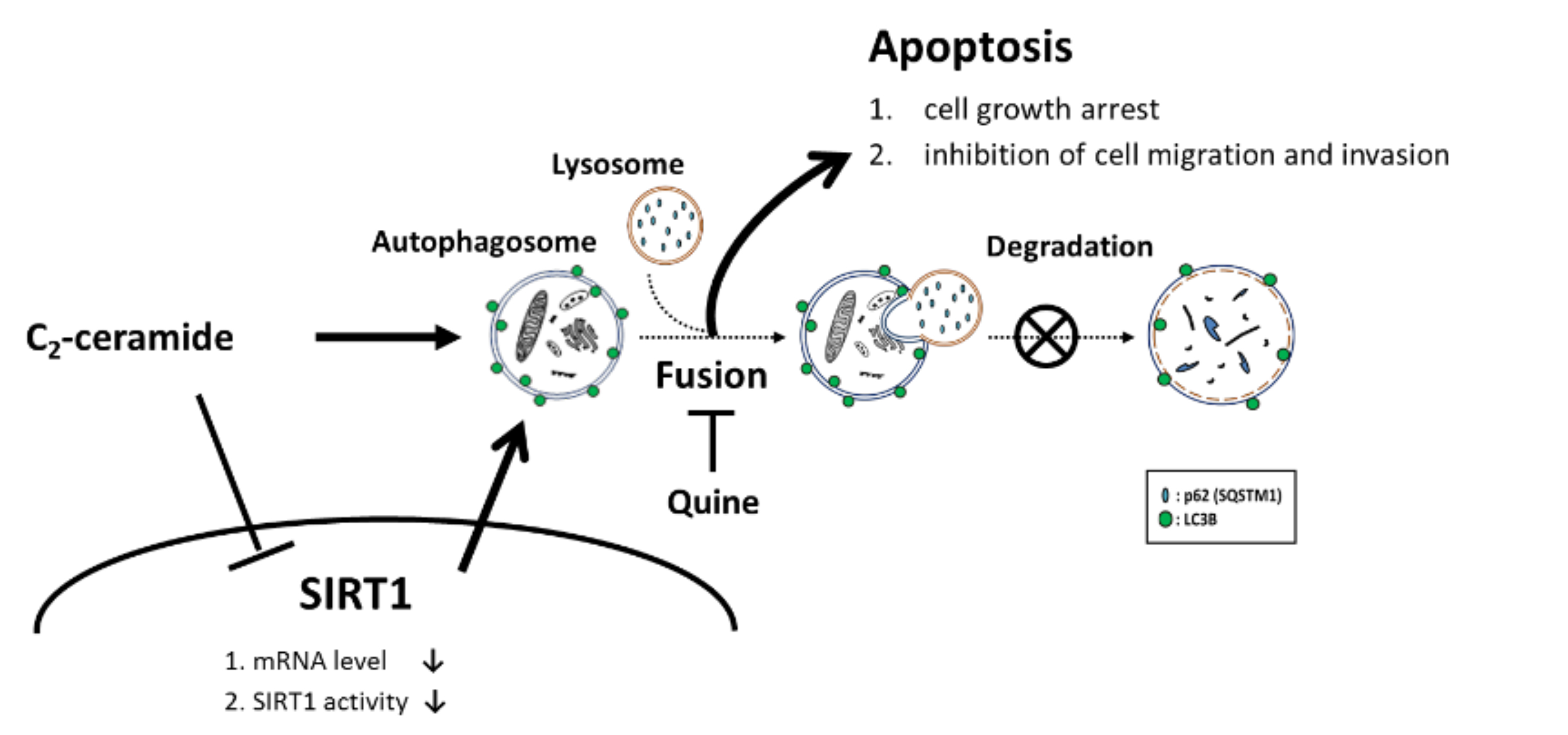
LC3 is a key regulator of autophagy that controls the major steps of this process, including the growth of autophagic membranes, the recognition of autophagic cargo, and the fusion of autophagosomes with lysosomes [
20,
21]. However, the increased expression of LC3 I/II was apparent in C
2-ceramide and CQ cotreated NSCLC cells. We speculate that the mechanism may involve the downregulation of SIRT1 induced by C
2-ceramide. SIRT1 is a NAD-dependent class III histone deacetylase that plays major roles in regulating gene expression, DNA damage repair, metabolism, tumor development, aging, and autophagy [
22]. Treatment with C
2-ceramide alone decreased SIRT1 expression in the nucleus, and cotreatment further reduced its expression. It has been reported that during autophagy, SIRT1 binds to endogenous LC3 and induces deacetylation, resulting in LC3 activation and autophagy progression [
23]. SIRT1 plays an important role in regulating the LC3 nucleus-cytoplasm shuttle. The absence of SIRT1 prevents LC3 from interacting with the nuclear protein DOR and affects the LC3 acetylation-deacetylation cycle and redistribution [
22]. These phenomena could contribute to the abnormal initiation of autophagy in our case. However, the detailed mechanism requires further investigation.
There is increasing evidence regarding the tyrosine kinase Src and its association with autophagy regulation. Tyrosine kinase inhibitors (TKIs) act as prototypes of target therapy and are used to treat various types of cancer in the clinic [
24,
25]. However, the efficacy of TKIs in inhibiting cancer progression and killing tumors is sometimes unsatisfactory and is accompanied by severe adverse effects. Previous studies have shown that patients who received TKIs for cancer treatment frequently experience adverse events, such as fatigue, diarrhea, etc. [
26,
27]. Thus, there is an urgent need to develop new drugs or treatment strategies. Our work showed that single treatment with C
2-ceramide induced autophagy accompanied by C-terminal Src family kinase (Csk) phosphorylation at Tyr527, which represents the inhibition of phosphorylation on downstream signaling [
28]. Generally, Src family kinases (SFKs) are activated by phosphorylation at Tyr416, resulting in the trans-phosphorylation of a cascade pathway leading to cell differentiation, proliferation, and survival. Unlike SFK activation, the phosphorylation of Csk deactivates SFKs during regular cellular processes. In addition, the combined treatment of C
2-ceramide and CQ induced the degradation of Src total protein and inhibited its activation via phosphorylation in both NSCLC cell lines. Interestingly, it was reported that an autophagy modulator could be used as a cotreatment with TKIs to induce additional cytotoxicity or an anti-cancer effect. Considering the inhibitory role of C
2-ceramide in the AKT pathway and the activation of Csk, C
2-ceramide could serve as an effective anti-cancer drug with potential to inhibit the PI3K/AKT/mTOR/p70S6K axis as an Src inhibitor, which is harmful to cell survival.
Taken together, our results suggest that single treatment with C2-ceramide-induced autophagy in NSCLC cells by inhibiting the translocation of SIRT1 into the nucleus to bind to LC3. Moreover, combined treatment with C2-ceramide and the autophagy inhibitor CQ enhanced cytotoxicity and autophagy accompanied by Src pathway inhibition. These results prove that a sublethal dose of C2-ceramide could be used with a lower risk, fewer adverse effects to achieve the same or better results and remarkable selectivity from tumor/normal cells through combined treatment. The current study sheds light on the combined usage of ceramide compounds and autophagy inhibitors in chemotherapy and translational medicine.
4. Materials and Methods
4.1. Reagents
Dulbecco’s modified Eagle’s medium (DMEM) was purchased from HyClone (Logan, UT, USA). Fetal bovine serum (FBS) was purchased from Gibco (Grand Island, NY, USA). Dimethyl sulfoxide (DMSO), trypan blue, penicillin G and streptomycin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against p-Src, Src, p-SIRT1, SIRT1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were purchased from Calbiochem (La Jolla, CA, USA). Antibodies against LAMP2 and LC3 I/II was purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-mouse and anti-rabbit IgG peroxidase-conjugated secondary antibodies were purchased from KPL (Gaithersburg, MD, USA).
4.2. Cell Culture
Human NSCLC H460 and H1299 cells were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were maintained in DMEM supplemented with 8% FBS, 2 mM glutamine, and antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin) at 37 °C in a humidified atmosphere of 5% CO2.
4.3. Cytotoxicity Assay
The survival rate of H460 and H1299 cells incubated with the compounds at the indicated concentrations were determined using the MTT Cell Proliferation Assay Kit according to the manufacturer’s instructions (Thermo Fisher, Carlsbad, CA, USA). Briefly, 1 × 105 cells were seeded and treated with the indicated concentrations of C2-ceramide (Sigma-Aldrich) and/or 10 µM CQ (Sigma-Aldrich) for 24 and 48 h. After incubation, 10 µL of 12 mM MTT stock solution was added, and the cells were incubated for 4 h at 37 °C. Then, 50 µL of DMSO was added and mixed for absorbance measurement at 540 nm.
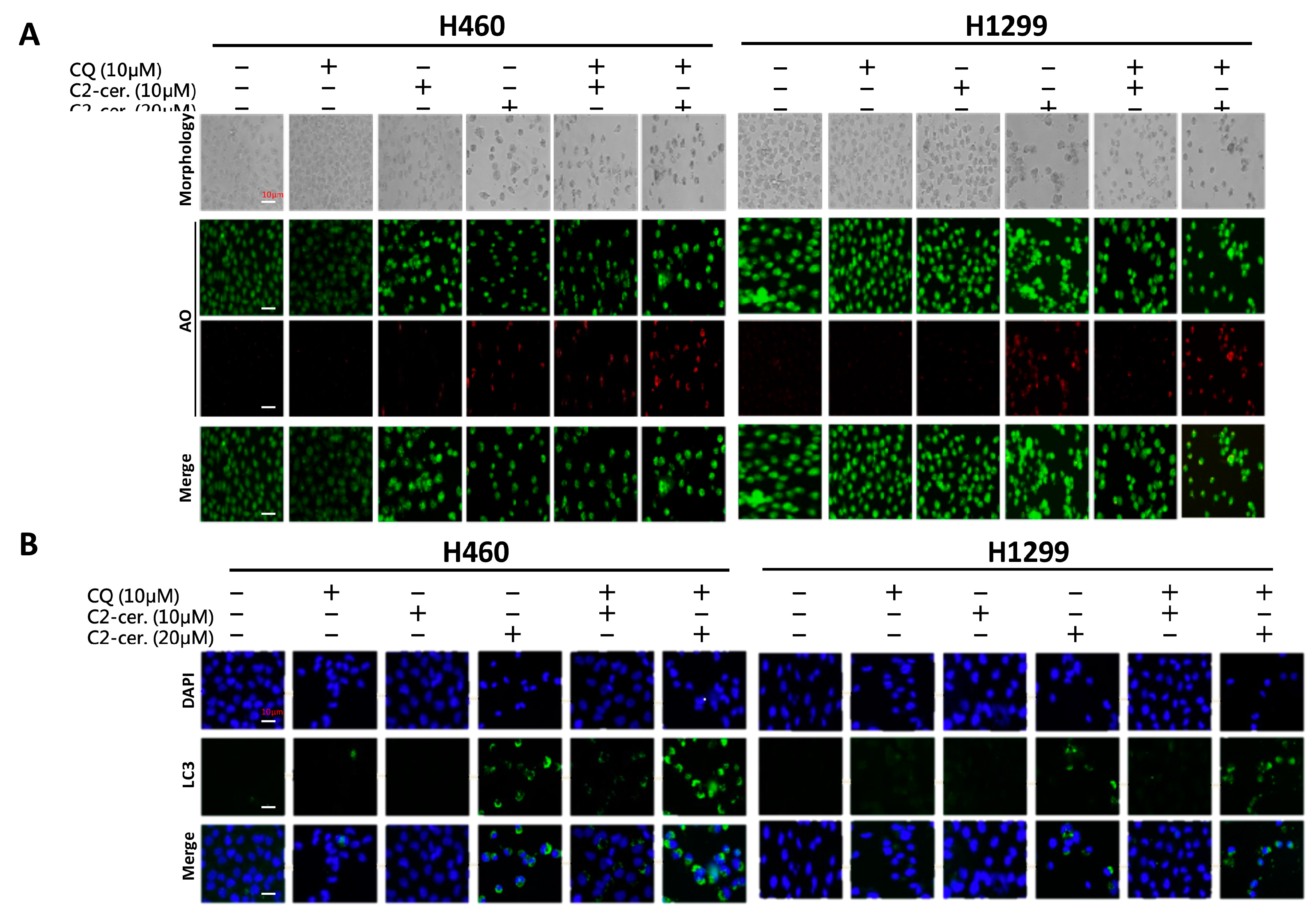
4.4. Acridine Orange (AO) Staining for Autophagy Determination
Acridine orange (AO) staining was performed according to a previously reported procedure [
29]. Briefly, 1 × 10
5 cells were seeded and treated with the indicated concentrations of C
2-ceramide and/or CQ for 24 h. Cells were then stained with 1 μg/mL AO (2.7 μM) in complete culture medium and incubated for 15 min at room temperature. Autophagy was detected using an immunofluorescence microscope for green (total cell) and red (AO-positive cell) light determination. Cell morphology was examined by using bright-field (BF) microscopy.
4.5. Immunofluorescence Staining
Assessment of protein distribution, including SIRT1, LC3, LAMP2, and SQSTM1, in both NSCLC cell lines was performed by employing immunofluorescent techniques. C2-Ceramide/CQ-treated cells were incubated in the presence of a primary monoclonal antibody against the protein of interest followed by incubation with an Alexa 594-conjugated (ex 594 nm/em 618 nm) secondary antibody. DAPI (4′,6-diamidino-2-phenylindole) was used as a counterstain to identify the nucleus, and cells were observed on a Leica immunofluorescence microscope (Leica Microsystems, Wetzlar, Germany). The samples were pretreated with 0.54% KCl and fixed with an acetic acid-methanol mixture (3:1) on glass slides. Fluorescent images were captured with an SD200 SpectraCube system (Applied Spectral Imaging, Migdal Ha’Emek, Israel) and mounted onto a Leica microscope. Digital images were optimized for image resolution (final resolution 300 dpi), brightness, and contrast using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA, USA). Images were not altered in any way, e.g., by removing or adding image details.
4.6. Cell Wound Healing Assay
Approximately 3 × 105 NSCLC cells were seeded onto a 12-well plate and grown to 100% confluence. Culture monolayers were scratched using a pipette tip to create a clean 1-mm-wide wound area. Cells were then incubated with phosphate buffered saline (PBS) (vehicle control), C2-ceramide (10–50 µM) and CQ (10 µM) either alone or in combination. After further incubation for 24 h, the wound gaps were imaged and analyzed using TScratch software (CSE Lab, Zurich, Switzerland).
4.7. Transwell Cell Invasion Assay
Invasion assays were performed as described in our previous work with slight modifications using 8-µm pore Transwell® chambers (Greiner Bio-One, Frickenhausen, Germany). Control and C2-ceramide- (10–50 µM) and CQ (10 µM)-treated cells were cultured in triplicate at 5 × 104 cells/well in the upper inserts of a 24-well Transwell® culture plate. Next, cells were fixed for 5 min and stained with 0.1% w/v Giemsa. Cells that had invaded the lower inserts were counted by arbitrarily selecting five fields from each well. The experiments were repeated three times.
4.8. Zebrafish Xenograft Assay
The additive inhibitory effect of CQ/ C
2-ceramide on NSCLC cells was validated using zebrafish-based xenograft assay according to our previous study with minor modifications [
30]. The protocol of zebrafish assay was approved (KMU-IACUC-102222) by the Institutional Animal Care and Use Committee (IACUC) of Kaohsiung Medical University, Kaohsiung, Taiwan. Briefly, H1299 cells were labeled with a red fluorescence dye 1,1′-dioctadecyl-3,3,3′,3′-tetra-methylendocarboxyamine (DiI) and 2 days postfertilization (dpf) zebrafish embryo were transplanted with 200 cells/embryo. The embryos were then incubated in water with indicated treatments including 5 μM of CQ, C
2-ceramide or CQ/ C
2-ceramide for 24 h postinjection respectively. Afterward, the red fluorescence of tumor mass was captured and analyzed.
4.9. Flow Cytometry and Annexin V/PI Double Staining
The flow cytometry-based cell cycle assay was performed as previously described [
31]. Briefly, 5 × 10
5 cells were treated with 10 μM CQ alone or cotreated with the indicated concentrations of C
2-ceramide for 72 h. Cells were then washed twice with pre-chilled PBS and collected by centrifugation at 200
g for 5 min at 4 °C. Cells were centrifuged, resuspended in 1 mL of propidium iodide (PI) staining buffer (10 μg/mL RNase A, 50 μg/mL PI in PBS) and an Annexin V Staining Kit (PharMingen, San Diego, CA, USA) double stain, followed by incubation at 37 °C for 30 min. The cells were then analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA, USA), and the results were analyzed using WinMDI 2.8 software (written by Joseph Trotter, Scripps Research Institute, La Jolla, CA, USA) [
32].
4.10. Western Blotting and Nuclear Protein Separation
Western blotting was performed according to a previous study [
31]. A total of 1 × 10
6 cells were treated with C
2-ceramide, CQ or both for 24 h. Cells were then harvested for protein extraction. The cells were lysed, and 40 μg of sample protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto a nitrocellulose membrane (Pall Life Science, Ann Arbor, MI, USA). The membranes were blocked with 5% non-fat milk and washed with PBS-T buffer before incubation with the corresponding primary and secondary antibodies. The signals were visualized using an Enhanced Chemiluminescence (ECL) Detection Kit (Amersham, Piscataway, NJ, USA) and analyzed by Gel Pro v.4.0 software (Media Cybernetics, Silver Spring, MD, USA).
4.11. Statistical Analysis
Each value represents the mean ± SD of at least three independent experiments. One-way analysis of variance (ANOVA) and paired sample t-tests were used to evaluate treatment significance at different time points of each experiment. Statistical significance was set at p < 0.05.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






