Cucurbitacin D Reprograms Glucose Metabolic Network in Prostate Cancer


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
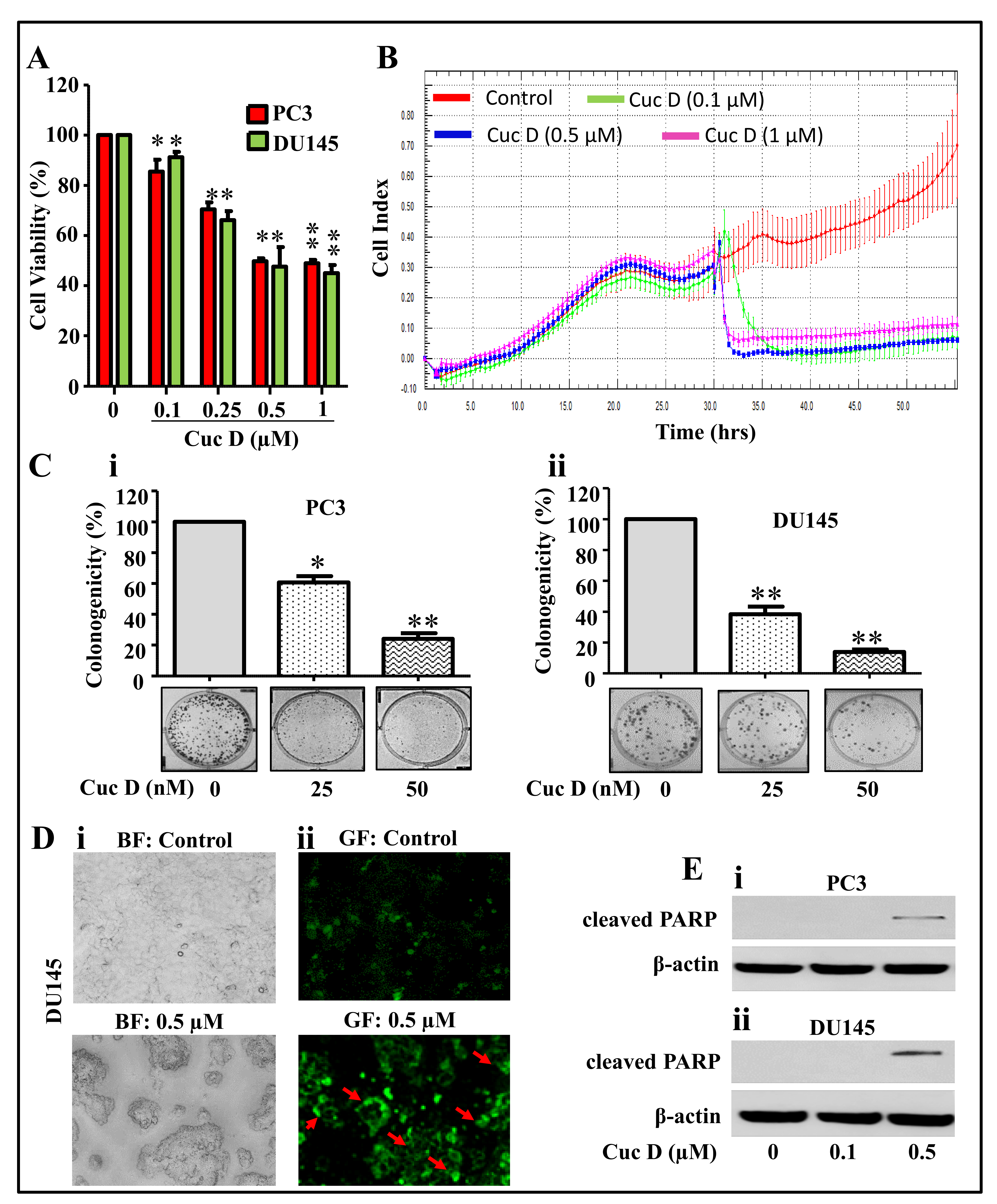
2.1. Cuc D Inhibits Growth and Induces Apoptosis Mediated Cell Death of PrCa Cells
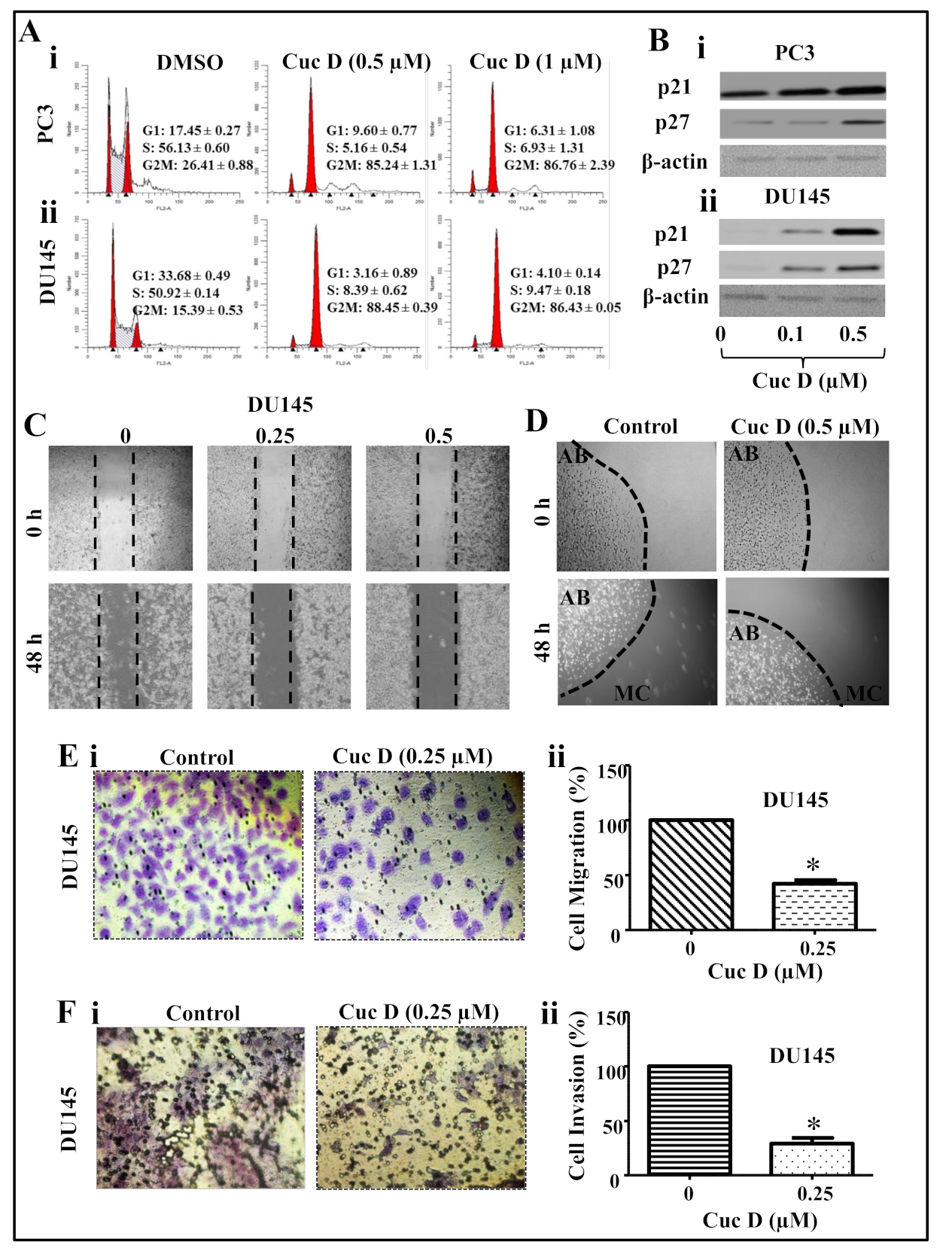
2.2. Cuc D Arrests Cell Cycle of PrCa Cells in G2/M Phase
2.3. Cuc D Inhibits The Migratory and Invasive Potential of PrCa Cells
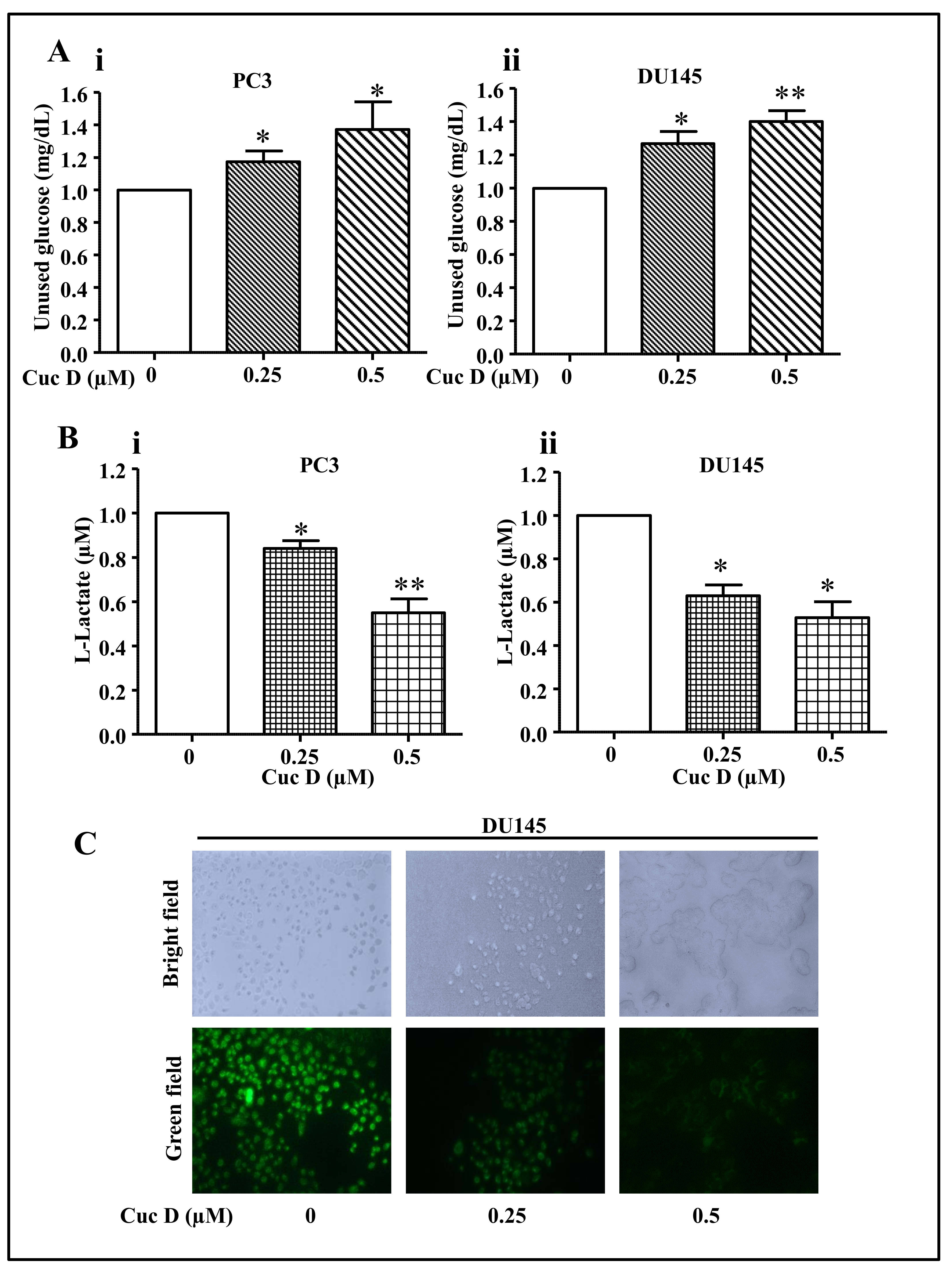
2.4. Cuc D Treatment Decreases Glucose Metabolism in PrCa Cells
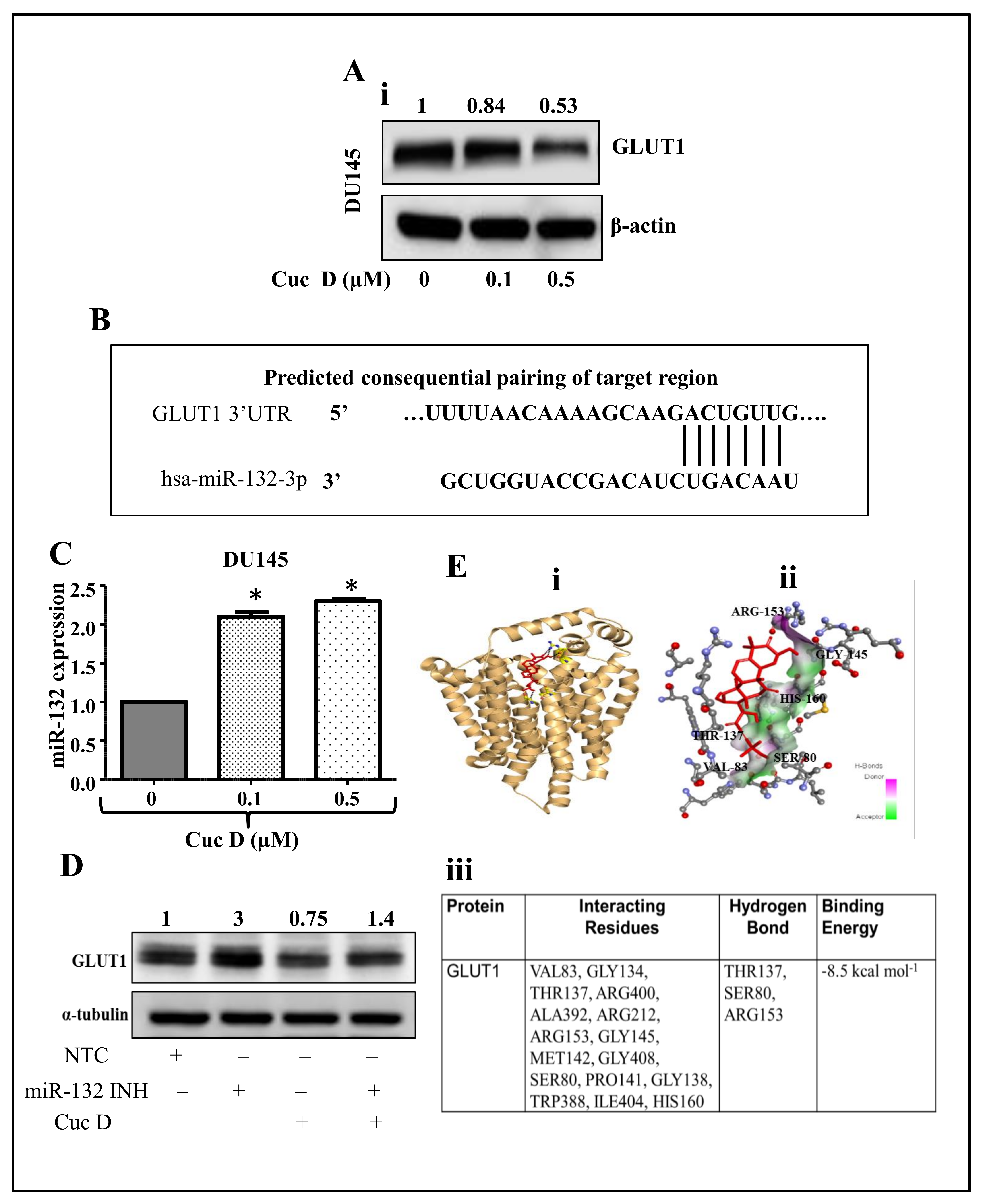
2.5. Cuc D Treatment Targets The GLUT1 Protein via Modulating miR-132 Expression
2.6. In Silico Studies Displays Cuc D Proficiently Bind with GLUT1
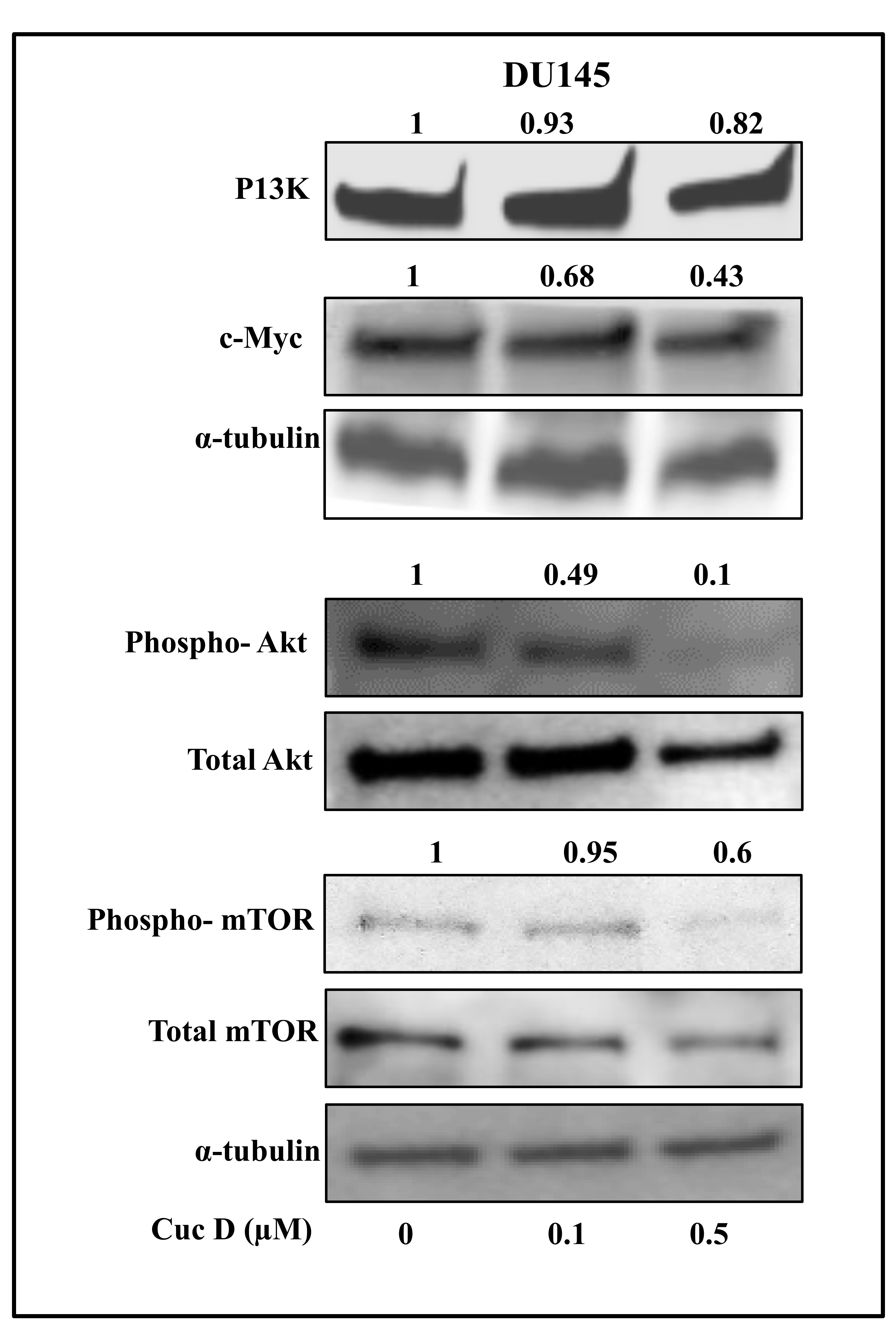
2.7. Cuc D Inhibits The Expression of Key Signaling Components Involved in Glucose Metabolism and Cell Survival in PrCa
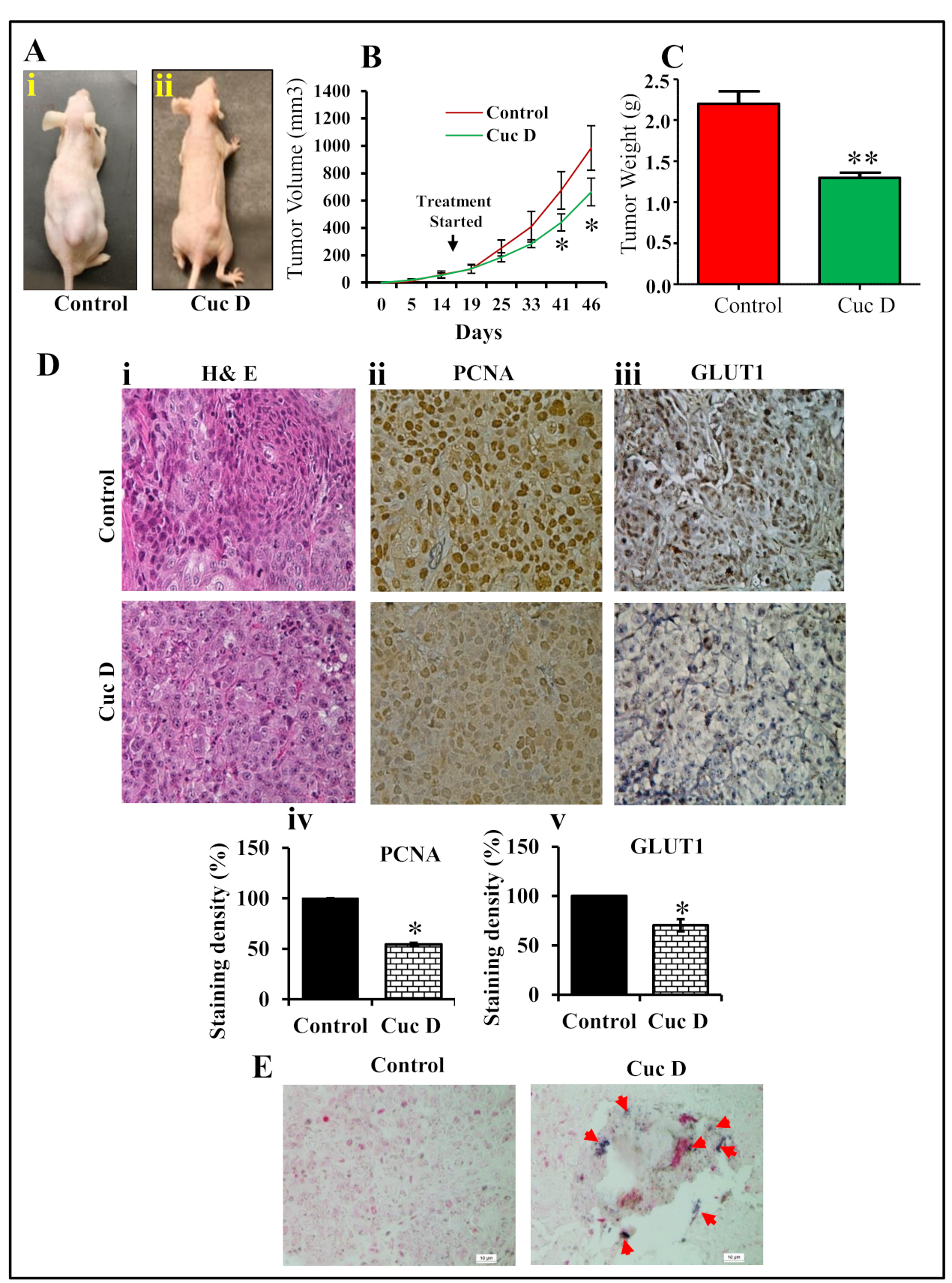
2.8. Cuc D Inhibits PrCa Cell Derived Xenograft Tumors in Athymic Nude Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Proliferation Assay
4.3. Cell Proliferation by xCELLigence Assay
4.4. Colony Formation Assay
4.5. Apoptosis Analysis
4.6. Cell Migration Assay
4.7. Agarose Bead Assay
4.8. Cell Invasion Assay
4.9. Cell Cycle Analysis
4.10. Western Blot Analysis
4.11. Isolation of RNA and PCR
4.12. Glucose and Lactate Assay
4.13. Xenograft Study
4.14. Immunohistochemistry (IHC)
4.15. In Situ Hybridization for miR-132
4.16. Molecular Docking
4.17. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Shukeir, N.; Potti, A.; Sircar, K.; Aprikian, A.; Goltzman, D.; Rabbani, S.A. Up-regulation of Wnt-1 and beta-catenin production in patients with advanced metastatic prostate carcinoma: Potential pathogenetic and prognostic implications. Cancer 2004, 101, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, B.B.; Ganju, A.; Sikander, M.; Kashyap, V.K.; Hafeez, Z.B.; Chauhan, N.; Malik, S.; Massey, A.E.; Tripathi, M.K.; Halaweish, F.T.; et al. Ormeloxifene Suppresses Prostate Tumor Growth and Metastatic Phenotypes via Inhibition of Oncogenic beta-catenin Signaling and EMT Progression. Mol. Cancer Ther. 2017, 16, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, R.N.; Karami-Tehrani, F.; Salami, S. Targeting prostate cancer cell metabolism: Impact of hexokinase and CPT-1 enzymes. Tumour Biol. 2015, 36, 2893–2905. [Google Scholar] [CrossRef]
- Oyama, N.; Akino, H.; Suzuki, Y.; Kanamaru, H.; Ishida, H.; Tanase, K.; Sadato, N.; Yonekura, Y.; Okada, K. FDG PET for evaluating the change of glucose metabolism in prostate cancer after androgen ablation. Nucl. Med. Commun. 2001, 22, 963–969. [Google Scholar] [CrossRef]
- Vaz, C.V.; Alves, M.G.; Marques, R.; Moreira, P.I.; Oliveira, P.F.; Maia, C.J.; Socorro, S. Androgen-responsive and nonresponsive prostate cancer cells present a distinct glycolytic metabolism profile. Int. J. Biochem. Cell Biol. 2012, 44, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Lakkis, C.L.; Laucirica, R.; Epner, D.E. Regulation of prostate cancer cell division by glucose. J. Cell. Physiol. 1999, 180, 431–438. [Google Scholar] [CrossRef]
- Kim, Y.S.; Milner, J.A. Bioactive food components and cancer-specific metabonomic profiles. J. Biomed. Biotechnol. 2011, 2011, 721213. [Google Scholar] [CrossRef]
- Rastogi, S.; Banerjee, S.; Chellappan, S.; Simon, G.R. Glut-1 antibodies induce growth arrest and apoptosis in human cancer cell lines. Cancer Lett. 2007, 257, 244–251. [Google Scholar] [CrossRef]
- Perez, A.; Ojeda, P.; Ojeda, L.; Salas, M.; Rivas, C.I.; Vera, J.C.; Reyes, A.M. Hexose transporter GLUT1 harbors several distinct regulatory binding sites for flavones and tyrphostins. Biochemistry 2011, 50, 8834–8845. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Chen, X.; Bergmeier, S.C. Novel inhibitors of basal glucose transport as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 2191–2194. [Google Scholar] [CrossRef]
- Jayaprakasam, B.; Seeram, N.P.; Nair, M.G. Anticancer and antiinflammatory activities of cucurbitacins from Cucurbita andreana. Cancer Lett. 2003, 189, 11–16. [Google Scholar] [CrossRef]
- Chen, J.C.; Chiu, M.H.; Nie, R.L.; Cordell, G.A.; Qiu, S.X. Cucurbitacins and cucurbitane glycosides: Structures and biological activities. Nat. Prod. Rep. 2005, 22, 386–399. [Google Scholar] [CrossRef]
- Thoennissen, N.H.; Iwanski, G.B.; Doan, N.B.; Okamoto, R.; Lin, P.; Abbassi, S.; Song, J.H.; Yin, D.; Toh, M.; Xie, W.D.; et al. Cucurbitacin B induces apoptosis by inhibition of the JAK/STAT pathway and potentiates antiproliferative effects of gemcitabine on pancreatic cancer cells. Cancer Res. 2009, 69, 5876–5884. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, M.; Shan, X.; Zhou, X.; Yang, J.; Wang, Y.; Li, J.; Deng, Y. Inhibitory effect of cucurbitacin E on pancreatic cancer cells growth via STAT3 signaling. J. Cancer Res. Clin. Oncol. 2010, 136, 603–610. [Google Scholar] [CrossRef]
- Sikander, M.; Hafeez, B.B.; Malik, S.; Alsayari, A.; Halaweish, F.T.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. Cucurbitacin D exhibits potent anti-cancer activity in cervical cancer. Sci. Rep. 2016, 6, 36594. [Google Scholar] [CrossRef]
- Shukla, S.; Shishodia, G.; Mahata, S.; Hedau, S.; Pandey, A.; Bhambhani, S.; Batra, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Aberrant expression and constitutive activation of STAT3 in cervical carcinogenesis: Implications in high-risk human papillomavirus infection. Mol. Cancer 2010, 9, 1476–4598. [Google Scholar] [CrossRef]
- Eidelman, E.; Twum-Ampofo, J.; Ansari, J.; Siddiqui, M.M. The Metabolic Phenotype of Prostate Cancer. Front. Oncol. 2017, 7, 131. [Google Scholar] [CrossRef]
- Qu, W.; Ding, S.M.; Cao, G.; Wang, S.J.; Zheng, X.H.; Li, G.H. miR-132 mediates a metabolic shift in prostate cancer cells by targeting Glut1. Febs Open Bio 2016, 6, 735–741. [Google Scholar] [CrossRef]
- Edinger, A.L.; Thompson, C.B. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell 2002, 13, 2276–2288. [Google Scholar] [CrossRef]
- Barata, J.T.; Silva, A.; Brandao, J.G.; Nadler, L.M.; Cardoso, A.A.; Boussiotis, V.A. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J. Exp. Med. 2004, 200, 659–669. [Google Scholar] [CrossRef]
- Xu, R.H.; Pelicano, H.; Zhang, H.; Giles, F.J.; Keating, M.J.; Huang, P. Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia 2005, 19, 2153–2158. [Google Scholar] [CrossRef]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef]
- Saito, S.; Furuno, A.; Sakurai, J.; Sakamoto, A.; Park, H.R.; Shin-Ya, K.; Tsuruo, T.; Tomida, A. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res. 2009, 69, 4225–4234. [Google Scholar] [CrossRef]
- Zhao, Y.; Coloff, J.L.; Ferguson, E.C.; Jacobs, S.R.; Cui, K.; Rathmell, J.C. Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J. Biol. Chem. 2008, 283, 36344–36353. [Google Scholar] [CrossRef]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef]
- Hardy, T.M.; Tollefsbol, T.O. Epigenetic diet: Impact on the epigenome and cancer. Epigenomics 2011, 3, 503–518. [Google Scholar] [CrossRef]
- Stefanska, B.; Karlic, H.; Varga, F.; Fabianowska-Majewska, K.; Haslberger, A. Epigenetic mechanisms in anti-cancer actions of bioactive food components—The implications in cancer prevention. Br. J. Pharm. 2012, 167, 279–297. [Google Scholar] [CrossRef]
- Gerhauser, C. Cancer chemoprevention and nutriepigenetics: State of the art and future challenges. Top. Curr. Chem. 2013, 329, 73–132. [Google Scholar]
- Phuah, N.H.; Nagoor, N.H. Regulation of microRNAs by natural agents: New strategies in cancer therapies. Biomed Res. Int. 2014, 2014, 804510. [Google Scholar] [CrossRef]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef]
- Roos, S.; Jansson, N.; Palmberg, I.; Saljo, K.; Powell, T.L.; Jansson, T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J. Physiol. 2007, 582, 449–459. [Google Scholar] [CrossRef]
- Plas, D.R.; Talapatra, S.; Edinger, A.L.; Rathmell, J.C.; Thompson, C.B. Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J. Biol. Chem. 2001, 276, 12041–12048. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef]
- He, X.; Gao, Q.; Qiang, Y.; Guo, W.; Ma, Y. Cucurbitacin E induces apoptosis of human prostate cancer cells via cofilin-1 and mTORC1. Oncol. Lett. 2017, 13, 4905–4910. [Google Scholar] [CrossRef]
- Yuan, G.; Yan, S.F.; Xue, H.; Zhang, P.; Sun, J.T.; Li, G. Cucurbitacin I induces protective autophagy in glioblastoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 10607–10619. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Wang, C.F.; Zhang, L.F. Cucurbitacin D impedes gastric cancer cell survival via activation of the iNOS/NO and inhibition of the Akt signalling pathway. Oncol. Rep. 2018, 39, 2595–2603. [Google Scholar] [CrossRef]
- Bartalis, J.; Halaweish, F.T. In vitro and QSAR studies of cucurbitacins on HepG2 and HSC-T6 liver cell lines. Bioorg. Med. Chem. 2011, 19, 2757–2766. [Google Scholar] [CrossRef]
- Limame, R.; Wouters, A.; Pauwels, B.; Fransen, E.; Peeters, M.; Lardon, F.; De Wever, O.; Pauwels, P. Comparative analysis of dynamic cell viability, migration and invasion assessments by novel real-time technology and classic endpoint assays. PLoS ONE 2012, 7, e46536. [Google Scholar] [CrossRef]
- Lee, C.M.; Fuhrman, C.B.; Planelles, V.; Peltier, M.R.; Gaffney, D.K.; Soisson, A.P.; Dodson, M.K.; Tolley, H.D.; Green, C.L.; Zempolich, K.A. Phosphatidylinositol 3-kinase inhibition by LY294002 radiosensitizes human cervical cancer cell lines. Clin. Cancer Res. 2006, 12, 250–256. [Google Scholar] [CrossRef]
- Naz, H.; Jameel, E.; Hoda, N.; Shandilya, A.; Khan, P.; Islam, A.; Ahmad, F.; Jayaram, B.; Hassan, M.I. Structure guided design of potential inhibitors of human calcium-calmodulin dependent protein kinase IV containing pyrimidine scaffold. Bioorg. Med. Chem. Lett. 2016, 26, 782–788. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.P.; Neumann, D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sikander, M.; Malik, S.; Chauhan, N.; Khan, P.; Kumari, S.; Kashyap, V.K.; Khan, S.; Ganju, A.; Halaweish, F.T.; Yallapu, M.M.; et al. Cucurbitacin D Reprograms Glucose Metabolic Network in Prostate Cancer. Cancers 2019, 11, 364. https://doi.org/10.3390/cancers11030364
Sikander M, Malik S, Chauhan N, Khan P, Kumari S, Kashyap VK, Khan S, Ganju A, Halaweish FT, Yallapu MM, et al. Cucurbitacin D Reprograms Glucose Metabolic Network in Prostate Cancer. Cancers. 2019; 11(3):364. https://doi.org/10.3390/cancers11030364
Chicago/Turabian StyleSikander, Mohammed, Shabnam Malik, Neeraj Chauhan, Parvez Khan, Sonam Kumari, Vivek Kumar Kashyap, Sheema Khan, Aditya Ganju, Fathi T. Halaweish, Murali M. Yallapu, and et al. 2019. "Cucurbitacin D Reprograms Glucose Metabolic Network in Prostate Cancer" Cancers 11, no. 3: 364. https://doi.org/10.3390/cancers11030364
APA StyleSikander, M., Malik, S., Chauhan, N., Khan, P., Kumari, S., Kashyap, V. K., Khan, S., Ganju, A., Halaweish, F. T., Yallapu, M. M., Jaggi, M., & Chauhan, S. C. (2019). Cucurbitacin D Reprograms Glucose Metabolic Network in Prostate Cancer. Cancers, 11(3), 364. https://doi.org/10.3390/cancers11030364