Liquid Biopsies in Lung Cancer: Four Emerging Technologies and Potential Clinical Applications

 , ,
, ,  , ,
, , 
Abstract
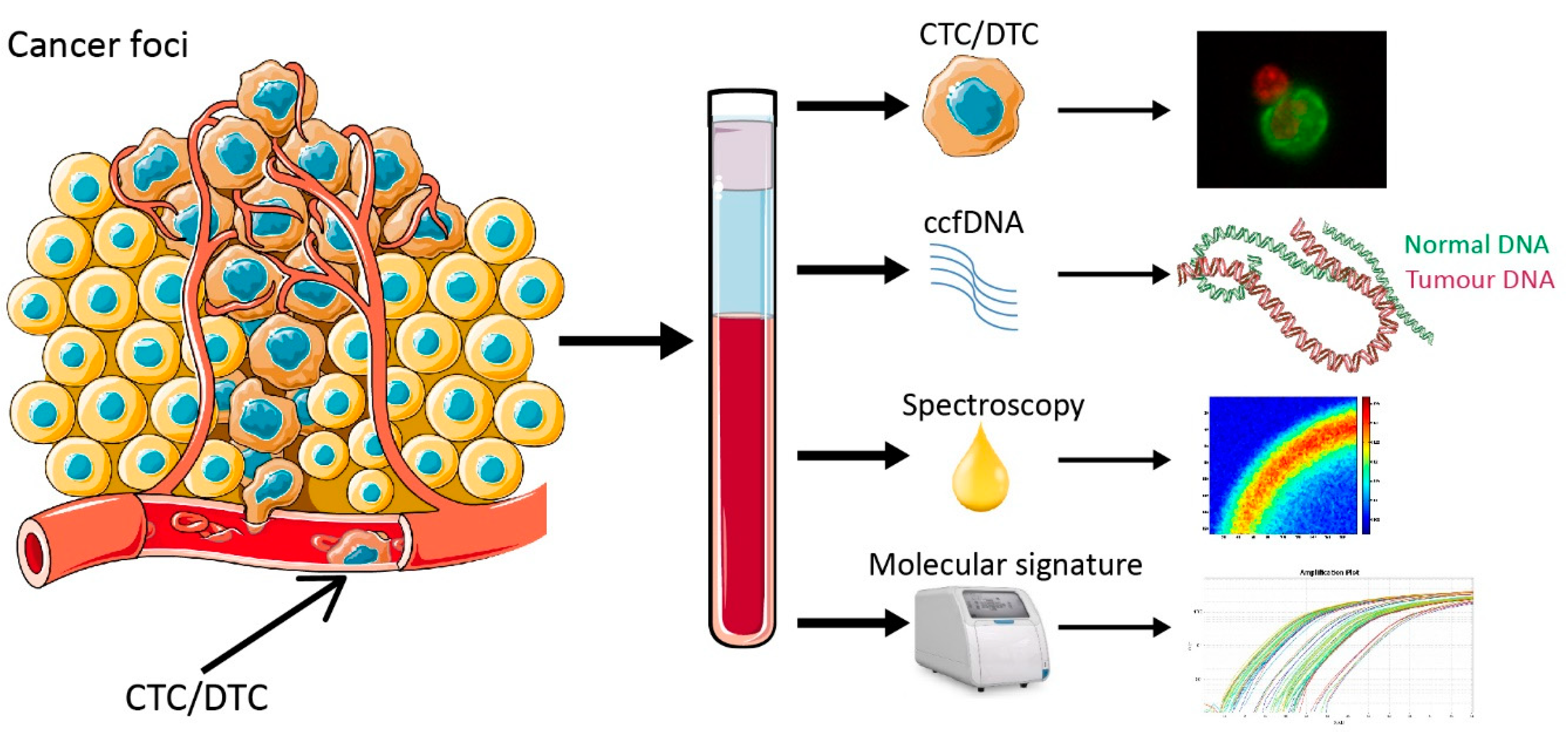
1. Introduction
2. Results
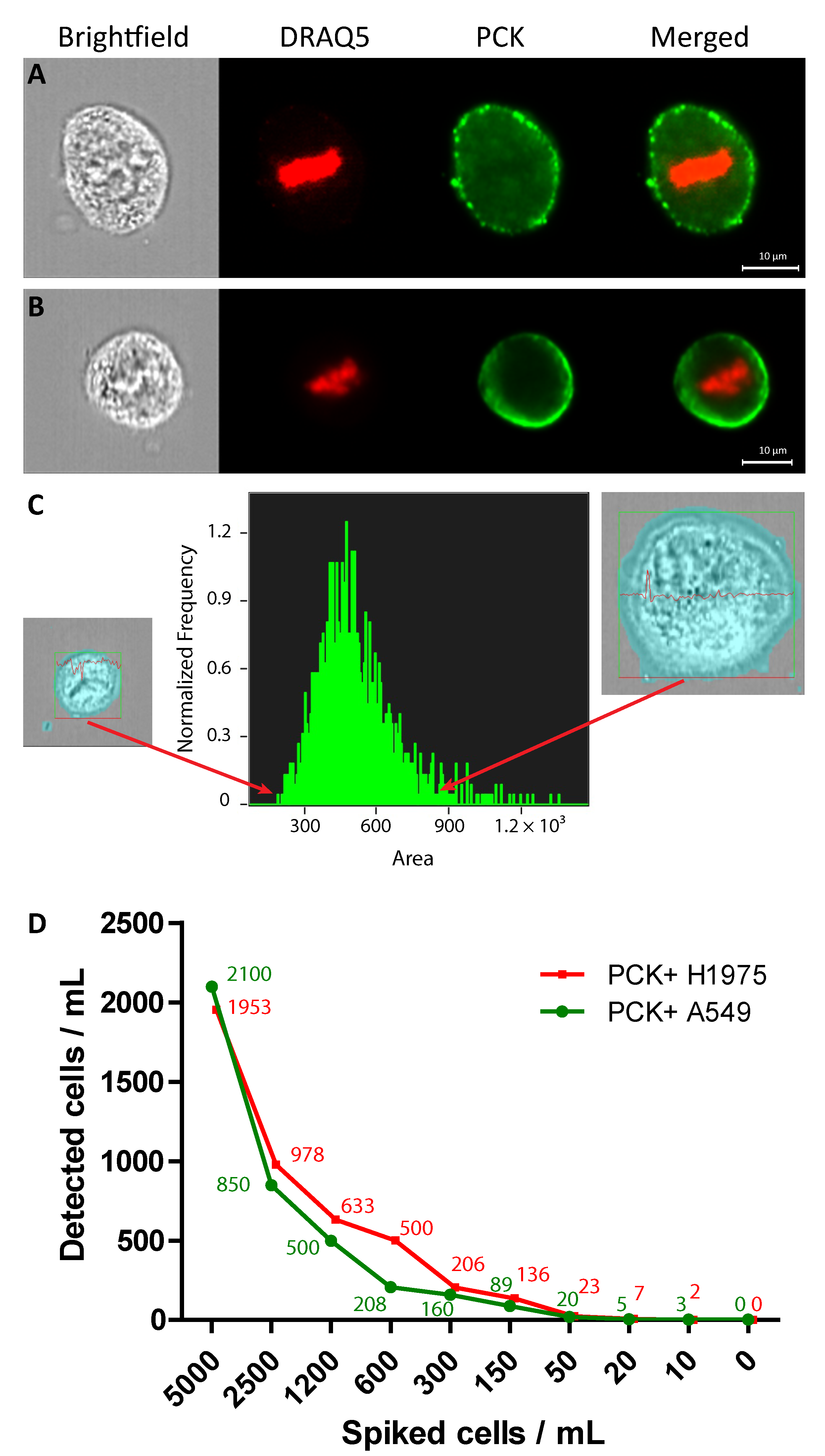
2.1. Identification of A549 and H1975 Lung Cancer Cells Mixed with Blood in Reconstruction Experiments Using AE1/AE3
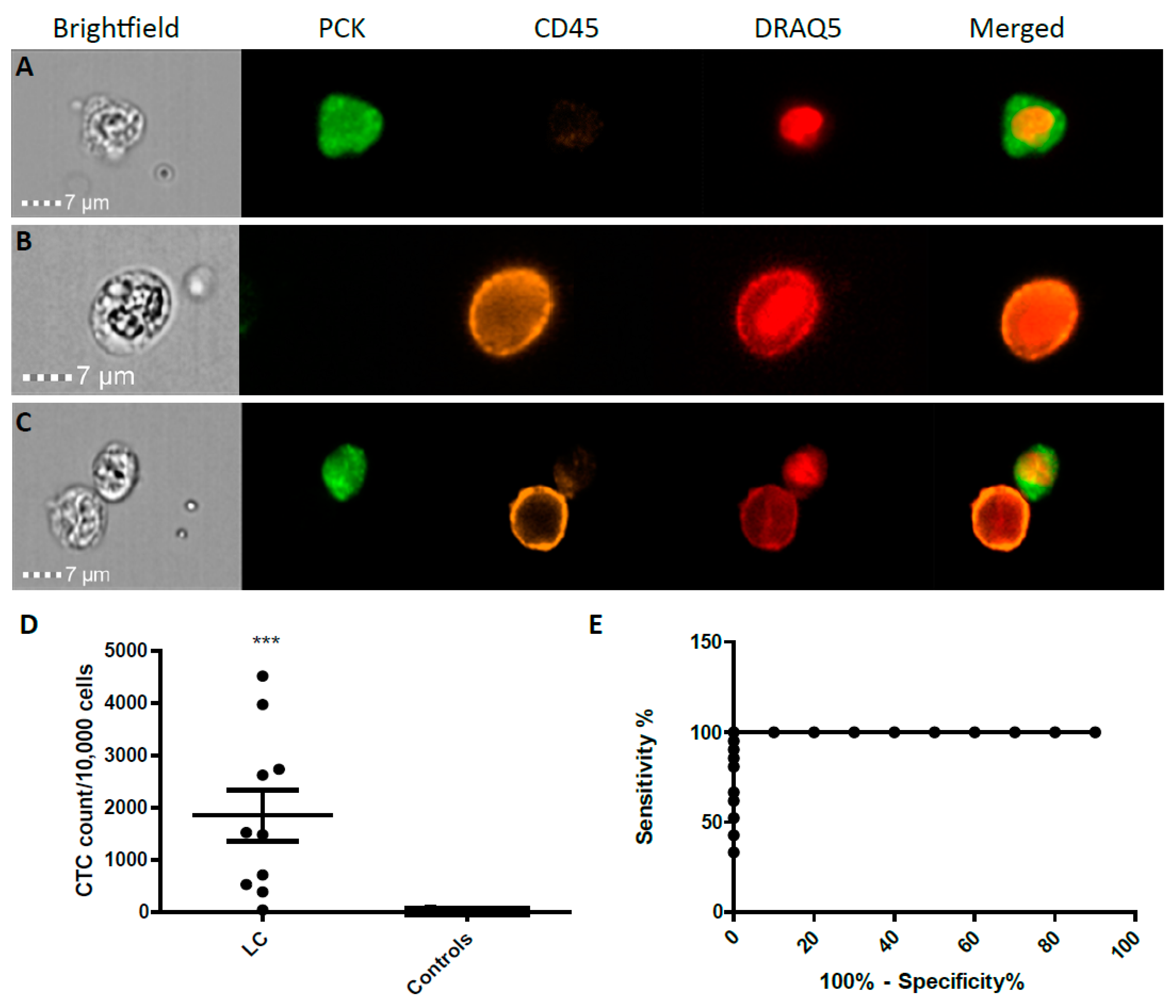
2.2. CTC Enumeration and Characterisation of LC Patients Using ImageStream™
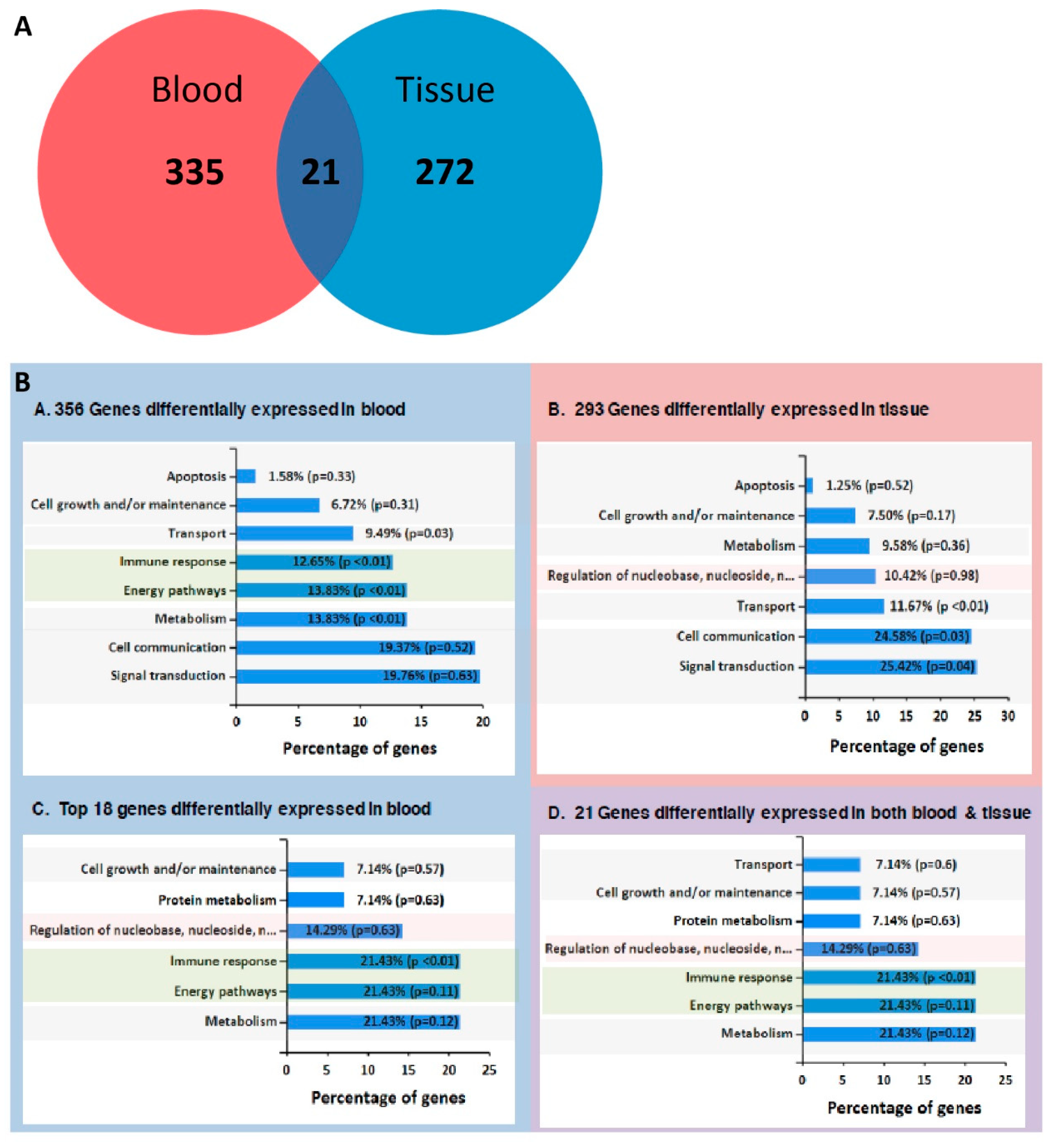
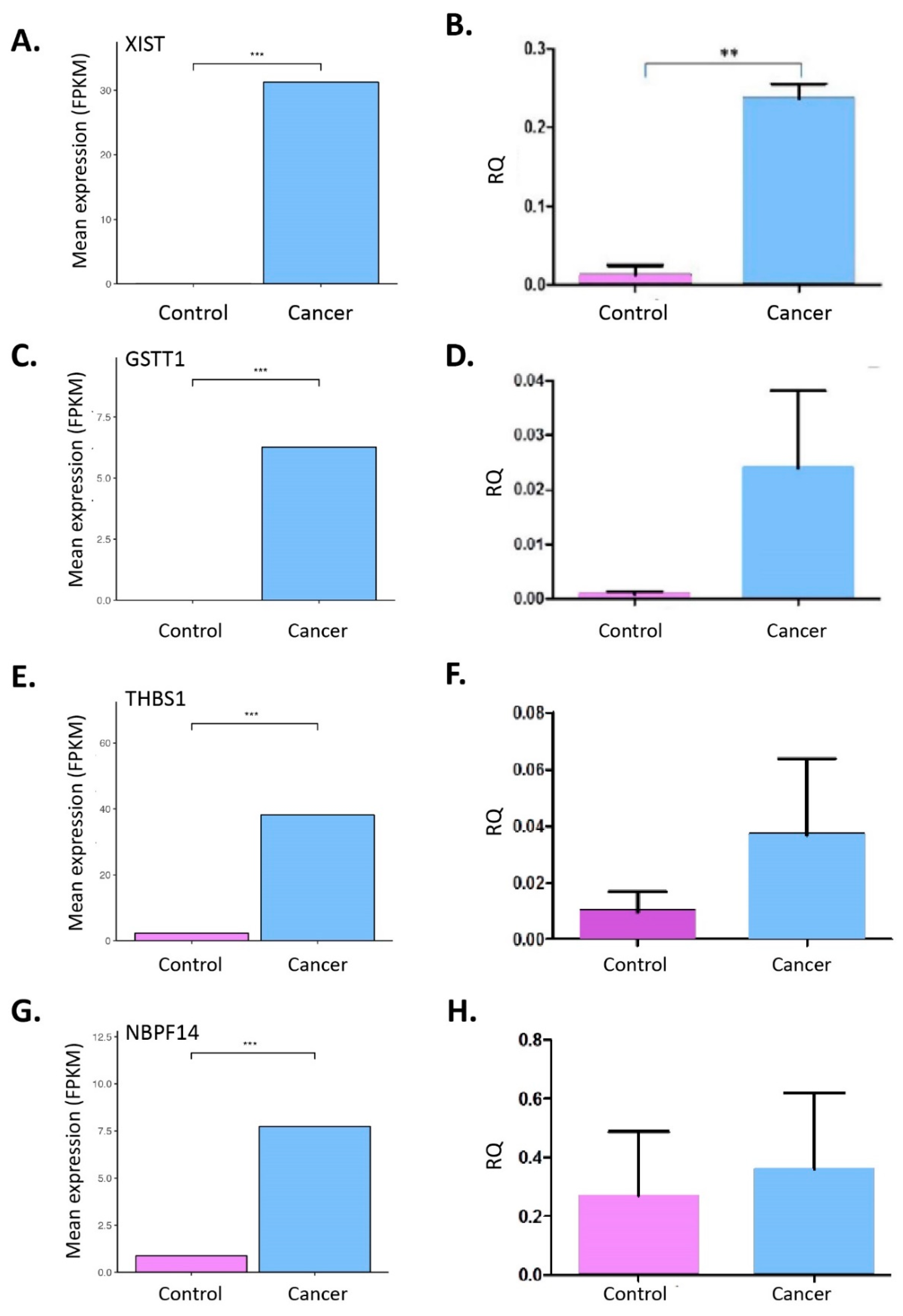
2.3. RNA-Seq from Whole Blood and Matched Tissues of LC Patients and Controls
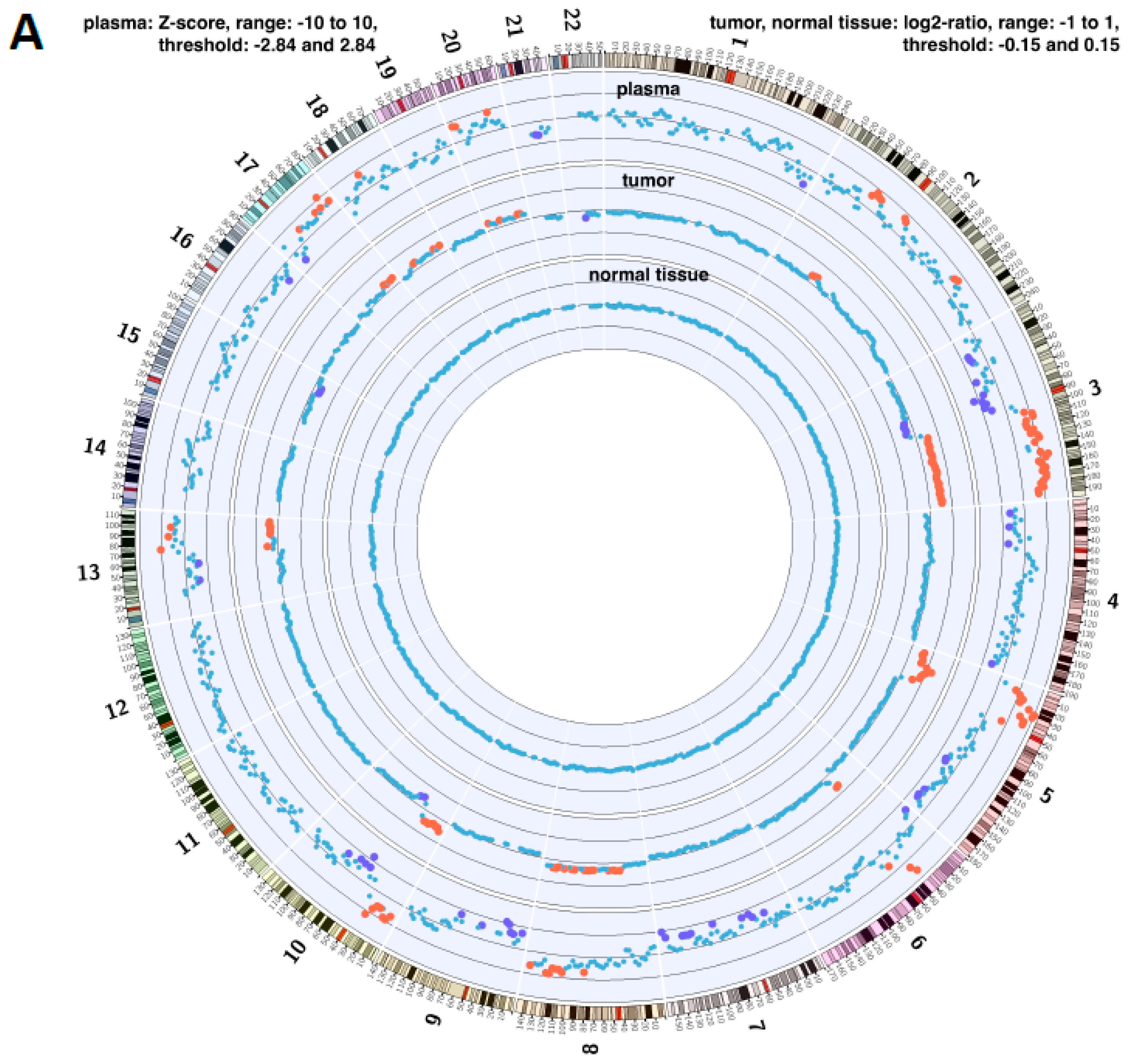
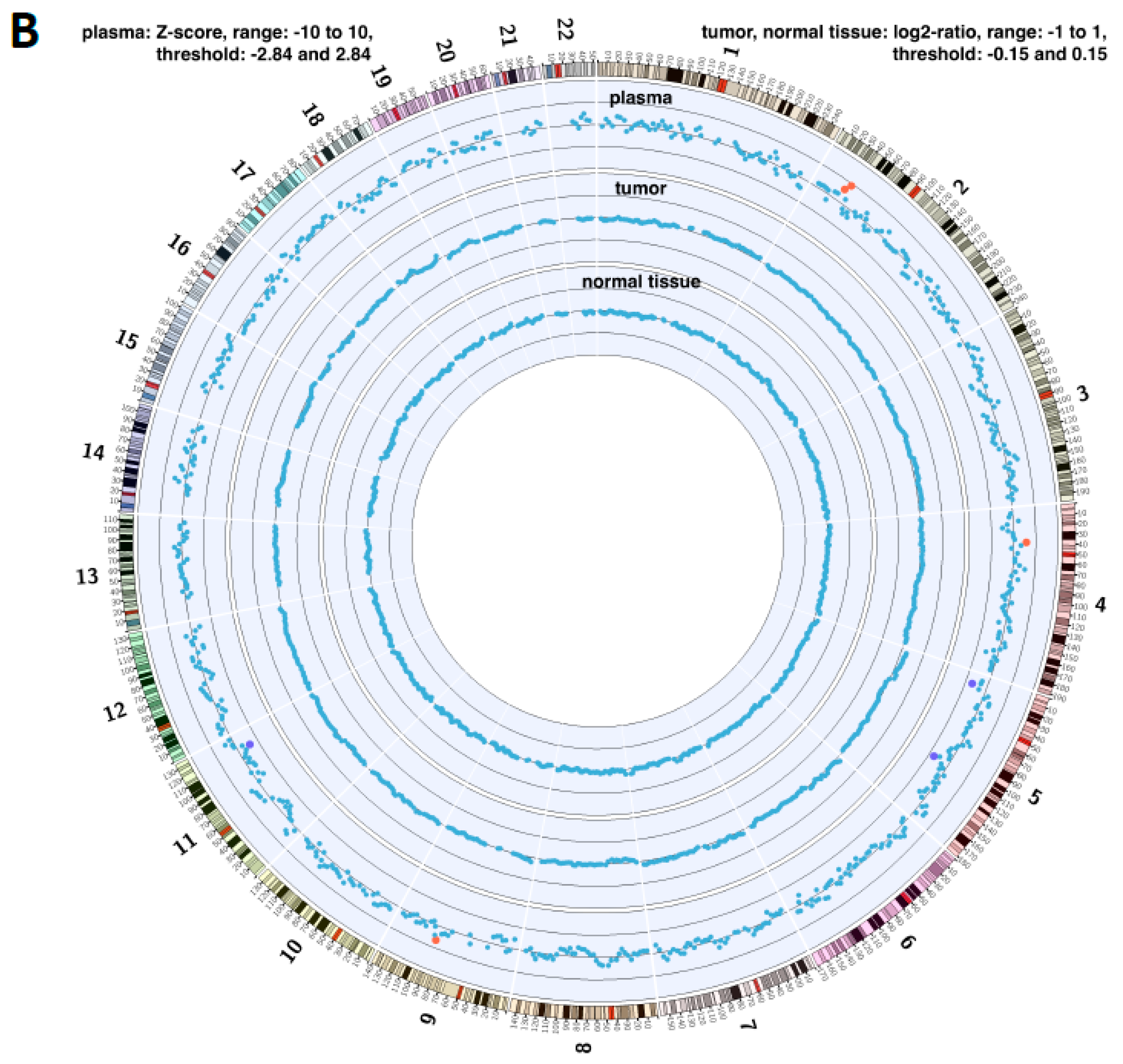
2.4. Copy Number Instability
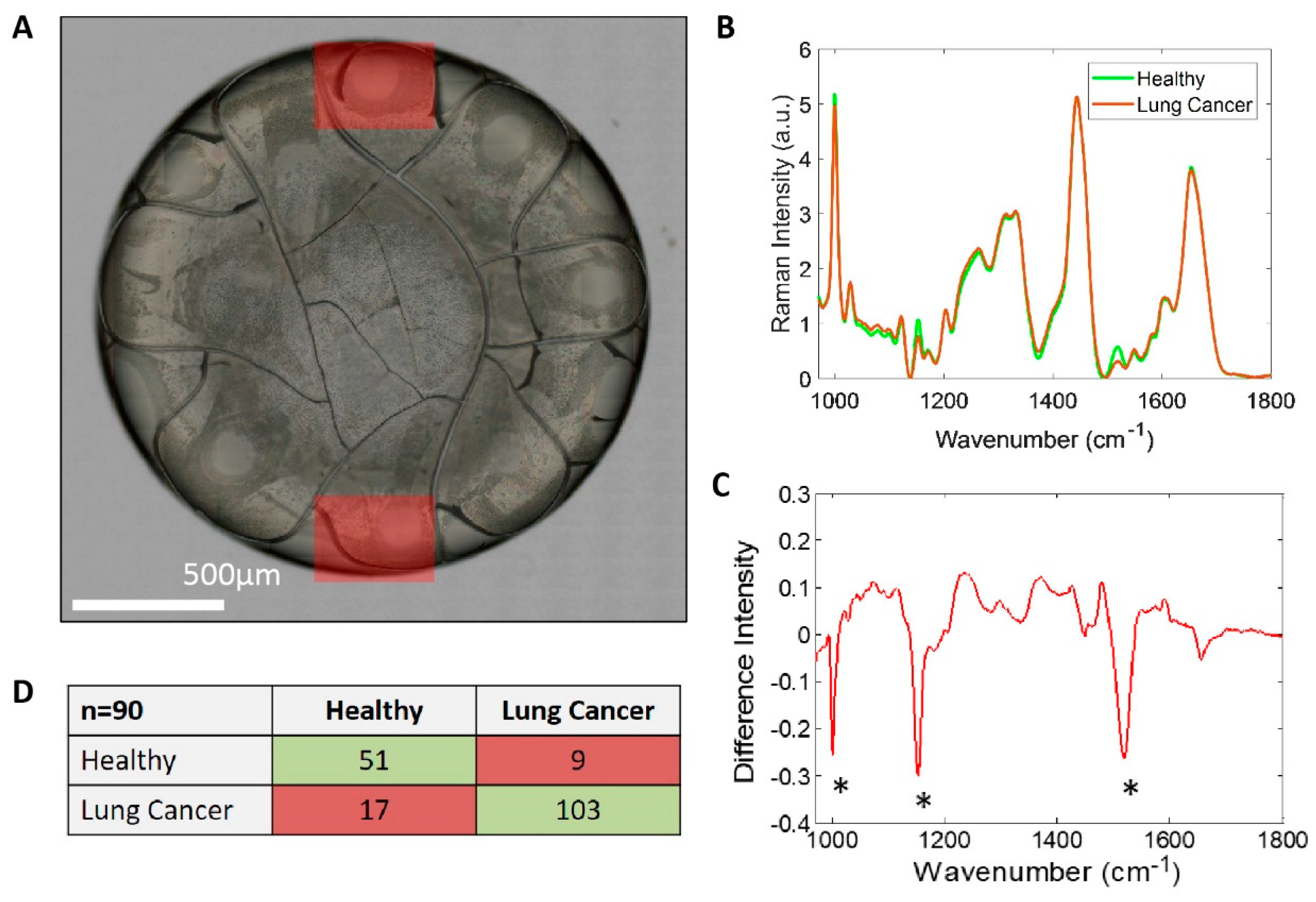
2.5. Raman Spectroscopy
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Preparation
4.2. Cell Lines
4.3. ImageStream Processing and Analysis
4.4. Raman Spectroscopy
4.5. Chromosome Number Instability Scoring and Analysis
4.6. RNA-Seq Processing and Analysis
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khoo, B.L.; Grenci, G.; Jing, T.; Lim, Y.B.; Lee, S.C.; Thiery, J.P.; Han, J.; Lim, C.T. Liquid biopsy and therapeutic response: Circulating tumor cell cultures for evaluation of anticancer treatment. Sci. Adv. 2016, 2, e1600274. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-L.; Chen, Y.-L.; Yang, S.-C.; Ho, C.-L.; Wei, F.; Wong, D.T.; Su, W.-C.; Lin, C.-C. Liquid biopsy genotyping in lung cancer: Ready for clinical utility? Oncotarget 2017, 8, 18590–18608. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Hayes, D.F.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Reuben, J.M.; Doyle, G.V.; Matera, J.; Allard, W.J.; Miller, M.C.; et al. Circulating tumor cells: A novel prognostic factor for newly diagnosed metastatic breast cancer. J. Clin. Oncol. 2005, 23, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.W.; Warkiani, M.E.; Khoo, B.L.; Li, Z.R.; Soo, R.A.; Tan, D.S.-W.; Lim, W.-T.; Han, J.; Bhagat, A.A.S.; Lim, C.T. Isolation and retrieval of circulating tumor cells using centrifugal forces. Sci. Rep. 2013, 3, 1259. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-M.; Lee, J.-Y.; Lee, J.-G.; Jeong, H.; Oh, J.-M.; Kim, Y.J.; Park, D.; Kim, M.S.; Lee, H.J.; Oh, J.H.; et al. Highly Efficient Assay of Circulating Tumor Cells by Selective Sedimentation with a Density Gradient Medium and Microfiltration from Whole Blood. Anal. Chem. 2012, 84, 7400–7407. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.G.; Hou, J.-M.; Sloane, R.; Lancashire, L.; Priest, L.; Nonaka, D.; Ward, T.H.; Backen, A.; Clack, G.; Hughes, A.; et al. Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and -independent approaches. J. Thorac. Oncol. 2012, 7, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Lim, A.R.; Ghajar, C.M. Circulating and disseminated tumor cells: Harbingers or initiators of metastasis? Mol. Oncol. 2017, 11, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Kapeleris, J.; Kulasinghe, A.; Warkiani, M.E.; Vela, I.; Kenny, L.; O’Byrne, K.; Punyadeera, C. The Prognostic Role of Circulating Tumor Cells (CTCs) in Lung Cancer. Front. Oncol. 2018, 8, 311. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, D.; Burnside, N.; Beeson, J.; Karteris, E.; Rice, A.; Anikin, V. Perioperative detection of circulating tumour cells in patients with lung cancer. Oncol. Lett. 2017, 14, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, D.; Freydina, D.V.; Freidin, M.B.; Leung, M.; Montero Fernandez, A.; Rice, A.; Nicholson, A.G.; Karteris, E.; Anikin, V.; Lim, E. Inertia based microfluidic capture and characterisation of circulating tumour cells for the diagnosis of lung cancer. Ann. Transl. Med. 2016, 4, 480. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Zhang, X.; Zhou, S.-L.; Cao, Y.; Huang, X.-W.; Fan, J.; Yang, X.-R.; Zhou, J. Plasma Circulating Cell-free DNA Integrity as a Promising Biomarker for Diagnosis and Surveillance in Patients with Hepatocellular Carcinoma. J. Cancer 2016, 7, 1798–1803. [Google Scholar] [CrossRef] [PubMed]
- Fournie, G.J.; Courtin, J.P.; Laval, F.; Chale, J.J.; Pourrat, J.P.; Pujazon, M.C.; Lauque, D.; Carles, P. Plasma DNA as a marker of cancerous cell death. Investigations in patients suffering from lung cancer and in nude mice bearing human tumours. Cancer Lett. 1995, 91, 221–227. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, D.Y.; Aladag, Z.; Felicien, M.I.; Hall, M.; Beeson, J.; Asadi, N.; Gidron, Y.; Karteris, E.; Anikin, V.B. Prognostic value of the DNA integrity index in patients with malignant lung tumors. Oncotarget 2018, 9, 21281–21288. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lung Cancer Incidence Statistics; Cancer Research UK: London, UK, 2018.
- Wong, M.C.S.; Lao, X.Q.; Ho, K.-F.; Goggins, W.B.; Tse, S.L.A. Incidence and mortality of lung cancer: Global trends and association with socioeconomic status. Sci. Rep. 2017, 7, 14300. [Google Scholar] [CrossRef] [PubMed]
- Al-Kattan, K.; Sepsas, E.; Fountain, S.W.; Townsend, E.R. Disease recurrence after resection for stage I lung cancer. Eur. J. Cardiothorac. Surg. 1997, 12, 380–384. [Google Scholar] [CrossRef]
- Uramoto, H.; Tanaka, F. Recurrence after surgery in patients with NSCLC. Transl. Lung Cancer Res. 2014, 3, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Carnio, S.; Novello, S.; Papotti, M.; Loiacono, M.; Scagliotti, G.V. Prognostic and predictive biomarkers in early stage non-small cell lung cancer: Tumor based approaches including gene signatures. Transl. Lung Cancer Res. 2013, 2, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D. Update on small cell carcinoma and its differentiation from squamous cell carcinoma and other non-small cell carcinomas. Mod. Pathol. 2012, 25, S18–S30. [Google Scholar] [CrossRef] [PubMed]
- Ogle, L.F.; Orr, J.G.; Willoughby, C.E.; Hutton, C.; McPherson, S.; Plummer, R.; Boddy, A.V.; Curtin, N.J.; Jamieson, D.; Reeves, H.L. Imagestream detection and characterisation of circulating tumour—A liquid biopsy for hepatocellular carcinoma? J. Hepatol. 2016, 65, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Mazurenka, M.; Behrendt, L.; Meinhardt-Wollweber, M.; Morgner, U.; Roth, B. Development of a combined OCT-Raman probe for the prospective in vivo clinical melanoma skin cancer screening. Rev. Sci. Instrum. 2017, 88, 105103. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, J.; Jiang, T.; Li, X.; Zhao, C.; Su, C.; Zhou, C. Predictive and prognostic value of folate receptor-positive circulating tumor cells in small cell lung cancer patients treated with first-line chemotherapy. Oncotarget 2017, 8, 49044–49052. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.G.; Sloane, R.; Priest, L.; Lancashire, L.; Hou, J.-M.; Greystoke, A.; Ward, T.H.; Ferraldeschi, R.; Hughes, A.; Clack, G.; et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Murata, Y.; Oki, Y.; Sugiyama, T.; Kusumoto, S.; Ishida, H.; Shirai, T.; Nakashima, M.; Yamaoka, T.; Okuda, K.; et al. Relationship of circulating tumor cells to the effectiveness of cytotoxic chemotherapy in patients with metastatic non-small-cell lung cancer. Oncol. Res. 2012, 20, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Farace, F.; Massard, C.; Vimond, N.; Drusch, F.; Jacques, N.; Billiot, F.; Laplanche, A.; Chauchereau, A.; Lacroix, L.; Planchard, D.; et al. A direct comparison of CellSearch and ISET for circulating tumour-cell detection in patients with metastatic carcinomas. Br. J. Cancer 2011, 105, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.; Morris, K.; Pritchard, S.; Kirwan, C.C. Challenges in enumeration of CTCs in breast cancer using techniques independent of cytokeratin expression. PLoS ONE 2017, 12, e0175647. [Google Scholar] [CrossRef] [PubMed]
- Jie, X.-X.; Zhang, X.-Y.; Xu, C.-J. Epithelial-to-mesenchymal transition, circulating tumor cells and cancer metastasis: Mechanisms and clinical applications. Oncotarget 2017, 8, 81558–81571. [Google Scholar] [CrossRef] [PubMed]
- Bitting, R.L.; Boominathan, R.; Rao, C.; Kemeny, G.; Foulk, B.; Garcia-Blanco, M.A.; Connelly, M.; Armstrong, A.J. Development of a method to isolate circulating tumor cells using mesenchymal-based capture. Methods 2013, 64, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.G.; Metcalf, R.L.; Carter, L.; Brady, G.; Blackhall, F.H.; Dive, C. Molecular analysis of circulating tumour cells—Biology and biomarkers. Nat. Rev. Clin. Oncol. 2014, 11, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Kenmotsu, H.; Koh, Y.; Yoshino, T.; Yoshikawa, T.; Naito, T.; Takahashi, T.; Murakami, H.; Nakamura, Y.; Tsuya, A.; et al. Size-Based Isolation of Circulating Tumor Cells in Lung Cancer Patients Using a Microcavity Array System. PLoS ONE 2013, 8, e67466. [Google Scholar] [CrossRef] [PubMed]
- Dent, B.M.; Ogle, L.F.; Donnell, R.L.O.; Hayes, N.; Malik, U.; Curtin, N.J.; Boddy, A.V.; Plummer, E.R.; Edmondson, R.J.; Reeves, H.L.; et al. Circulating Tumour Cells from Patients with Oesophageal, Hepatocellular, Thyroid and Ovarian. Cancers 2016, 216, 206–216. [Google Scholar]
- Najjar, F.; Alammar, M.; Bachour, M.; Almalla, N.; Altahan, M.; Alali, A.; Al-Massarani, G. Predictive and prognostic value of circulating endothelial cells in non-small cell lung cancer patients treated with standard chemotherapy. J. Cancer Res. Clin. Oncol. 2015, 141, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Weakley, S.M.; Wang, H.; Yao, Q.; Chen, C. Expression and function of a large non-coding RNA gene XIST in human cancer. World J. Surg. 2011, 35, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Xue, S.; Zeng, B.; Qiu, D. lncRNA SNHG5 is associated with poor prognosis of bladder cancer and promotes bladder cancer cell proliferation through targeting p27. Oncol. Lett. 2018, 15, 1924–1930. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Sun, C.-C.; Gong, C. Long noncoding RNA XIST acts as an oncogene in non-small cell lung cancer by epigenetically repressing KLF2 expression. Biochem. Biophys. Res. Commun. 2016, 478, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xue, Y.; Wang, P.; Liu, X.; Ma, J.; Zheng, J.; Li, Z.; Li, Z.; Cai, H.; Liu, Y. Knockdown of long non-coding RNA XIST increases blood–tumor barrier permeability and inhibits glioma angiogenesis by targeting miR-137. Oncogenesis 2017, 6, e303. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shen, Q.; Zhang, X.; Yang, C.; Cui, S.; Sun, Y.; Wang, L.; Fan, X.; Xu, S. The long non-coding RNA XIST controls non-small cell lung cancer proliferation and invasion by modulating miR-186-5p. Cell. Physiol. Biochem. 2017, 41, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Raman, C.V.; Krishnan, K.S. A New Type of Secondary Radiation. Nature 1928, 121, 501. [Google Scholar] [CrossRef]
- Deegan, R.D.; Bakajin, O.; Dupont, T.F.; Huber, G.; Nagel, S.R.; Witten, T.A. Capillary flow as the cause of ring stains from dried liquid drops. Nature 1997, 389, 827. [Google Scholar] [CrossRef]
- Filik, J.; Stone, N. Drop coating deposition Raman spectroscopy of protein mixtures. Analyst 2007, 132, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Filik, J.; Stone, N. Analysis of human tear fluid by Raman spectroscopy. Anal. Chim. Acta 2008, 616, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.E. Advantageous Uses of Mass Spectrometry for the Quantification of Proteins. Int. J. Proteom. 2013, 2013, 219452. [Google Scholar] [CrossRef] [PubMed]
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 11, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Fiedor, J.; Fiedor, L.; Haessner, R.; Scheer, H. Cyclic endoperoxides of beta-carotene, potential pro-oxidants, as products of chemical quenching of singlet oxygen. Biochim. Biophys. Acta 2005, 1709, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Nishino, H.; Tokuda, H.; Murakoshi, M.; Satomi, Y.; Masuda, M.; Onozuka, M.; Yamaguchi, S.; Takayasu, J.; Tsuruta, J.; Okuda, M.; et al. Cancer prevention by natural carotenoids. Biofactors 2000, 13, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Winnard, P.T.J.; Zhang, C.; Vesuna, F.; Kang, J.W.; Garry, J.; Dasari, R.R.; Barman, I.; Raman, V. Organ-specific isogenic metastatic breast cancer cell lines exhibit distinct Raman spectral signatures and metabolomes. Oncotarget 2017, 8, 20266–20287. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.; Kendall, C.; Stone, N.; Notingher, I. Raman spectroscopy for medical diagnostics—From in-vitro biofluid assays to in-vivo cancer detection. Adv. Drug Deliv. Rev. 2015, 89, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.D.; Lewis, K.E.; Ghosal, R.; Bayliss, S.; Lloyd, A.J.; Wills, J.; Godfrey, R.; Kloer, P.; Mur, L.A.J. Evaluation of FTIR Spectroscopy as a diagnostic tool for lung cancer using sputum. BMC Cancer 2010, 10, 640. [Google Scholar] [CrossRef] [PubMed]
- Holliday, S.F.; Kane-Gill, S.; Empey, P.E.; Buckley, M.; Smithburger, P. Interpatient Variability in Dexmedetomidine Response: A Survey of the Literature. Sci. World J. 2014, 2014, 805013. [Google Scholar] [CrossRef] [PubMed]
- Munzone, E.; Botteri, E.; Sandri, M.T.; Esposito, A.; Adamoli, L.; Zorzino, L.; Sciandivasci, A.; Cassatella, M.C.; Rotmensz, N.; Aurilio, G.; et al. Prognostic value of circulating tumor cells according to immunohistochemically defined molecular subtypes in advanced breast cancer. Clin. Breast Cancer 2012, 12, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Hennecke, S.; Bornemann-Kolatzki, K.; Urnovitz, H.B.; Neumann, S.; Ströbel, P.; Kaup, F.-J.; Brenig, B.; Schütz, E. Genome Aberrations in Canine Mammary Carcinomas and Their Detection in Cell-Free Plasma DNA. PLoS ONE 2013, 8, e75485. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Beck, J.; Braun, D.P.; Bornemann-Kolatzki, K.; Barilla, H.; Cubello, R.; Quan, W.J.; Sangal, A.; Khemka, V.; Waypa, J.; et al. Tumor Cell-Free DNA Copy Number Instability Predicts Therapeutic Response to Immunotherapy. Clin. Cancer Res. 2017, 23, 5074–5081. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Hood, B.L.; Zhao, T.; Conrads, T.P.; Sun, M.; Gopalakrishnan, V.; Grover, H.; Day, R.S.; Weissfeld, J.L.; Wilson, D.O.; et al. Lung cancer serum biomarker discovery using label-free liquid chromatography-tandem mass spectrometry. J. Thorac. Oncol. 2011, 6, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.T.; Gaifulina, R.; Isabelle, M.; Dorney, J.; Woods, M.L.; Lloyd, G.R.; Lau, K.; Rodriguez-Justo, M.; Kendall, C.; Stone, N.; et al. Mirrored stainless steel substrate provides improved signal for Raman spectroscopy of tissue and cells. J. Raman Spectrosc. 2017, 48, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Butler, H.J.; Ashton, L.; Bird, B.; Cinque, G.; Curtis, K.; Dorney, J.; Esmonde-White, K.; Fullwood, N.J.; Gardner, B.; Martin-Hirsch, P.L.; et al. Using Raman spectroscopy to characterize biological materials. Nat. Protoc. 2016, 11, 664–687. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.; Kendall, C.; Smith, J.; Crow, P.; Barr, H. Raman spectroscopy for identification of epithelial cancers. Faraday Discuss. 2004, 126, 141–183. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, J.; Kendall, C.; Shepherd, N.; Barr, H.; Stone, N. Evaluation of linear discriminant analysis for automated Raman histological mapping of esophageal high-grade dysplasia. J. Biomed. Opt. 2010, 15, 66015. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.A.; Beck, J.; Leu, M.; Oellerich, M.; Rave-Fränk, M.; Walson, P.D.; Schütz, E.; Canis, M. Cell-Free Plasma DNA for Disease Stratification and Prognosis in Head and Neck Cancer. Clin. Chem. 2018, 64, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Pathan, M.; Keerthikumar, S.; Ang, C.-S.; Gangoda, L.; Quek, C.Y.J.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef] [PubMed]
- Gallart-Palau, X.; Serra, A.; Sze, S.K. Enrichment of extracellular vesicles from tissues of the central nervous system by PROSPR. Mol. Neurodegener. 2016, 11, 41. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pre-Operative Characteristics | n | Percentage |
|---|---|---|
| Male/Female | 6 | 46.2 |
| Age at surgery (median) | 70 years (range 34–79) | - |
| BMI (median) | 21.1 | - |
| Smoker (current or ex) | 11 | 84.6 |
| Diabetes | 3 | 23.1 |
| IHD | 5 | 38.5 |
| Surgical resection | ||
| Wedge | 2 | 15.4 |
| Segmentectomy | 1 | 7.7 |
| Lobectomy | 9 | 69.2 |
| Bilobectomy | 1 | 7.7 |
| Surgical approach | ||
| VATS | 4 | 30.8 |
| Thoracotomy | 9 | 69.2 |
| Stage (lung cancer patients) | ||
| I | 3 | 30.0 |
| II | 5 | 50.0 |
| III | 0 | 0.0 |
| IV | 2 | 20.0 |
| Histology | ||
| Lung squamous cell carcinoma | 2 | 15.4 |
| Lung adenocarcinoma | 8 | 61.5 |
| Metastatic adenocarcinoma | 3 | 23.1 |
| Survival | ||
| Dead | 6 | 46.2 |
| Alive | 7 | 53.8 |
| Median survival, months | 22.8 (range 0.6–24.8) | - |
| Pre-Operative Characteristics | n | Percentage |
|---|---|---|
| Male/Female | 12 | 60.0 |
| Age at surgery (median) | 70 years (range 34–79) | - |
| BMI (median) | 21.6 | - |
| Smoker (current or ex) | 15 | 75.0 |
| Diabetes | 3 | 15.0 |
| IHD | 5 | 25.0 |
| Surgical resection | ||
| Wedge | 2 | 10.0 |
| Segmentectomy | 1 | 5.0 |
| Lobectomy | 15 | 75.0 |
| Bilobectomy | 1 | 5.0 |
| Pneumonectomy | 1 | 5.0 |
| Surgical approach | ||
| VATS | 6 | 30.0 |
| Thoracotomy | 14 | 70.0 |
| Stage (lung cancer patients) | ||
| I | 3 | 15.0 |
| II | 9 | 45.0 |
| III | 2 | 10.0 |
| IV | 2 (10.0%) | |
| Histology | ||
| Lung squamous cell carcinoma | 8 | 40.0 |
| Lung adenocarcinoma | 8 | 40.0 |
| Metastasis | 4 | 20.0 |
| Survival | ||
| Dead | 7 | 35.0 |
| Alive | 13 | 65.0 |
| Median survival, months | 26.2 (range 0.6–28.9) | - |
| Control Characteristics | n | Percentage |
|---|---|---|
| Male/Female | 8 | 38.0 |
| Age at surgery (median) | 49 years (range 28–70) | - |
| Control type | ||
| Healthy volunteer | 18 | 86.0 |
| Bullectomy | 3 | 14.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chudasama, D.; Katopodis, P.; Stone, N.; Haskell, J.; Sheridan, H.; Gardner, B.; Urnovitz, H.; Schuetz, E.; Beck, J.; Hall, M.; et al. Liquid Biopsies in Lung Cancer: Four Emerging Technologies and Potential Clinical Applications. Cancers 2019, 11, 331. https://doi.org/10.3390/cancers11030331
Chudasama D, Katopodis P, Stone N, Haskell J, Sheridan H, Gardner B, Urnovitz H, Schuetz E, Beck J, Hall M, et al. Liquid Biopsies in Lung Cancer: Four Emerging Technologies and Potential Clinical Applications. Cancers. 2019; 11(3):331. https://doi.org/10.3390/cancers11030331
Chicago/Turabian StyleChudasama, Dimple, Periklis Katopodis, Nick Stone, Jennifer Haskell, Hannah Sheridan, Benjamin Gardner, Howard Urnovitz, Ekkehard Schuetz, Julia Beck, Marcia Hall, and et al. 2019. "Liquid Biopsies in Lung Cancer: Four Emerging Technologies and Potential Clinical Applications" Cancers 11, no. 3: 331. https://doi.org/10.3390/cancers11030331
APA StyleChudasama, D., Katopodis, P., Stone, N., Haskell, J., Sheridan, H., Gardner, B., Urnovitz, H., Schuetz, E., Beck, J., Hall, M., Barr, J., Sisu, C., Rice, A., Polychronis, A., Anikin, V., & Karteris, E. (2019). Liquid Biopsies in Lung Cancer: Four Emerging Technologies and Potential Clinical Applications. Cancers, 11(3), 331. https://doi.org/10.3390/cancers11030331