Tumor Neovascularization and Developments in Therapeutics

 , and
, and
Abstract
1. Introduction
2. Molecules Involved in Neovascularization
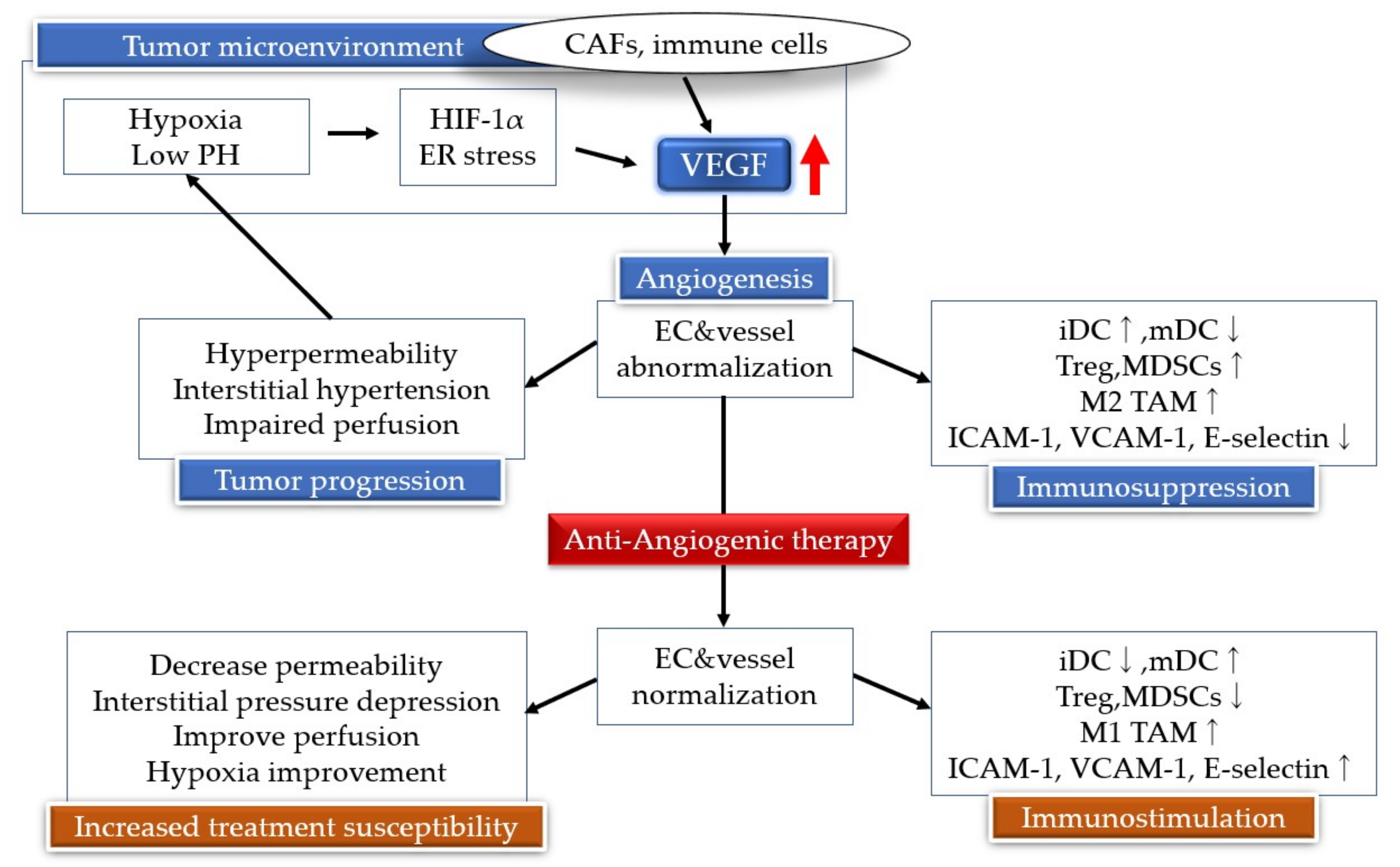
3. Characteristics of Angiogenic Tumor Vessels
4. Regulatory Mechanisms of Neovascularization
4.1. HIF-1α
4.2. Endoplasmic Reticulum Stress Signals
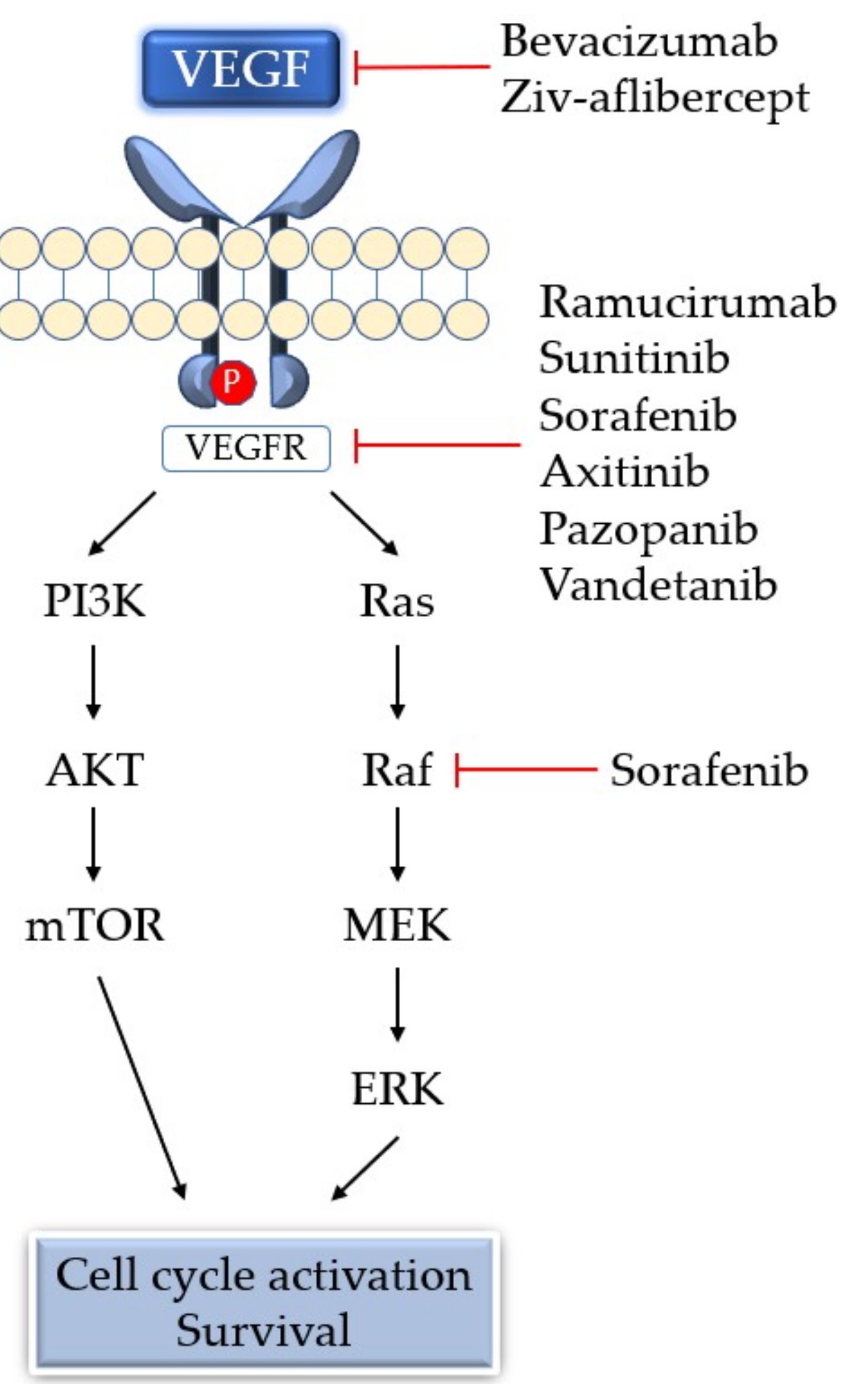
5. Antiangiogenic Therapy
6. Resistance Mechanism of Angiogenesis Inhibitors
7. Neovascularization and Immunity
8. Angiogenesis Inhibitors and Immunotherapy
9. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Tepper, O.M.; Capla, J.M.; Galiano, R.D.; Ceradini, D.J.; Callaghan, M.J.; Kleinman, M.E.; Gurtner, G.C. Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow-derived cells. Blood 2005, 105, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Kalluri, R. Cancer with disease. Nature 2004, 427, 787. [Google Scholar] [CrossRef] [PubMed]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Molecular Sciences Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Benjamin, L.E.; Francisco, S. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 6, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Snuderl, M.; Batista, A.; Kirkpatrick, N.D.; De Almodovar, C.R.; Riedemann, L.; Walsh, E.C.; Anolik, R.; Huang, Y.; Martin, J.D.; Kamoun, W.; et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 2013, 152, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef] [PubMed]
- Klein, D. The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front. Oncol. 2018, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing Tumor Microenvironment to Treat Cancer: Bench to Bedside to Biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Karaman, S.; Leppänen, V.-M.; Alitalo, K. Vascular endothelial growth factor signaling in development and disease. Development 2018, 145, dev151019. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Zarkada, G.; Wallgard, E.; Murtomäki, A.; Suchting, S.; Wirzenius, M.; Waltari, M.; Hellström, M.; Schomber, T.; Peltonen, R.; et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008, 454, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Meadows, K.N.; Bryant, P.; Pumiglia, K. Vascular Endothelial Growth Factor Induction of the Angiogenic Phenotype Requires Ras Activation. J. Biol. Chem. 2001, 276, 49289–49298. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Wu, W.; Mosteller, R.D.; Broek, D. Sphingosine kinase mediates vascular endothelial growth factor-induced activation of ras and mitogen-activated protein kinases. Mol. Cell. Biol. 2002, 22, 7758–7768. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamaguchi, S.; Chida, K.; Shibuya, M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 2001, 20, 2768–2778. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [PubMed]
- Fujio, Y.; Walsh, K. Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J. Biol. Chem. 1999, 274, 16349–16354. [Google Scholar] [CrossRef] [PubMed]
- Michell, B.J.; Griffiths, J.E.; Mitchelhill, K.I.; Rodriguez-Crespo, I.; Tiganis, T.; Bozinovski, S.; de Montellano, P.R.; Kemp, B.E.; Pearson, R.B. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 1999, 9, 845–848. [Google Scholar] [CrossRef]
- Murohara, T.; Horowitz, J.R.; Silver, M.; Tsurumi, Y.; Chen, D.; Sullivan, A.; Isner, J.M. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation 1998, 97, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): A dual regulator for angiogenesis. Angiogenesis 2006, 9, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Kubo, H.; Fujiwara, T. Involvement ofvascular endothelial growth factor receptor-3 in maintenance ofintegrity ofendothelial cell lining during tumor angiogenesis. Blood 2000, 96, 546–553. [Google Scholar] [PubMed]
- Parikh, S.M. The Angiopoietin-Tie2 Signaling Axis in Systemic Inflammation. J. Am. Soc. Nephrol. 2017, 28, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, K.; de Aos Scherpenseel, I.; Jain, S.K.; Presman, E.; Varticovski, L.; Varticovski, L. Role of PI 3-Kinase in Angiopoietin-1-Mediated Migration and Attachment-Dependent Survival of Endothelial Cells. Exp. Cell Res. 1999, 253, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Kim, H.G.; So, J.N.; Kim, J.H.; Kwak, H.J.; Koh, G.Y. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3’-Kinase/Akt signal transduction pathway. Circ. Res. 2000, 86, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.P.; Marron, M.B.; Brindle, N.P.J. The Antiinflammatory Endothelial Tyrosine Kinase Tie2 Interacts With a Novel Nuclear Factor-κB Inhibitor ABIN-2. Circ. Res. 2003, 92, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Sako, K.; Minami, T.; Noda, K.; Kim, H.Z.; Kodama, T.; Shibuya, M.; Takakura, N.; Koh, G.Y.; Mochizuki, N. Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat. Cell Biol. 2008, 10, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Wiegand, S.J.; Yancopoulos, G.D. New model of tumor angiogenesis: Dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. Oncogene 1999, 18, 5356–5362. [Google Scholar] [CrossRef] [PubMed]
- Nagy, N. Endothelial cells promote migration and proliferation of enteric neural crest cells via β1 integrin signaling. Dev. Biol. 2009, 330, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Kalinski, P. Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy; Springer International Publishing: New York, NY, USA, 2017; ISBN 331967577X. [Google Scholar]
- Tong, R.T.; Boucher, Y.; Kozin, S.V.; Winkler, F.; Hicklin, D.J.; Jain, R.K. Vascular Normalization by Vascular Endothelial Growth Factor Receptor 2 Blockade Induces a Pressure Gradient Across the Vasculature and Improves Drug Penetration in Tumors. Cancer Res. 2004, 64, 3731–3736. [Google Scholar] [CrossRef] [PubMed]
- Streit, M.; Riccardi, L.; Velasco, P.; Brown, L.F.; Hawighorst, T.; Bornstein, P.; Detmar, M. Thrombospondin-2: A potent endogenous inhibitor of tumor growth and angiogenesis. Proc. Natl. Acad. Sci. USA 1999, Dec 21, 14888–14893. [Google Scholar] [CrossRef]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Fitzgerald, G.; Soro-Arnaiz, I.; De Bock, K. The Warburg Effect in Endothelial Cells and its Potential as an Anti-angiogenic Target in Cancer. Front. Cell Dev. Biol. 2018, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Inai, T.; Mancuso, M.; Hashizume, H.; Baffert, F.; Haskell, A.; Baluk, P.; Hu-lowe, D.D.; Shalinsky, D.R.; Thurston, G.; Yancopoulos, G.D.; et al. Inhibition of Vascular Endothelial Growth Factor (VEGF) Signaling in Cancer Causes Loss of Endothelial Fenestrations, Regression of Tumor Vessels, and Appearance of Basement Membrane Ghosts. Am. J. Pathol. 2004, 165, 35–52. [Google Scholar] [CrossRef]
- Ferland-McCollough, D.; Slater, S.; Richard, J.; Reni, C.; Mangialardi, G. Pericytes, an overlooked player in vascular pathobiology. Pharmacol. Ther. 2017, 171, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Kawamoto, T.; Ohga, N.; Akiyama, K.; Hida, Y.; Shindoh, M. Altered angiogenesis in the tumor microenvironment. Pathol. Int. 2011, 61, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.-C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Kidoya, H.; Sakimoto, S.; Wakabayashi, T.; Takakura, N. Identification and characterization of a resident vascular stem/progenitor cell population in preexisting blood vessels. EMBO J. 2012, 31, 842–855. [Google Scholar] [CrossRef] [PubMed]
- Ohmura-Kakutani, H.; Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Kawamoto, T.; Iida, J.; Shindoh, M.; Tsuchiya, K.; Shinohara, N.; et al. Identification of tumor endothelial cells with high aldehyde dehydrogenase activity and a highly angiogenic phenotype. PLoS ONE 2014, 9, e113910. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Wakabayashi, T.; Kidoya, H.; Muramatsu, F.; Takara, K.; Eino, D.; Yamane, K.; Iba, T.; Takakura, N. Endothelial side population cells contribute to tumor angiogenesis and antiangiogenic drug resistance. Cancer Res. 2016, 76, 3200–3210. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A.; et al. Tumor endothelial cells acquire drug resistance by MDR1 up-regulation via VEGF signaling in tumor microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, M.; Ohga, N.; Akiyama, K.; Hida, Y.; Maishi, N.; Towfik, A.M.; Inoue, N.; Shindoh, M.; Hida, K. Hypoxia-induced reactive oxygen species cause chromosomal abnormalities in endothelial cells in the tumor microenvironment. PLoS ONE 2013, 8, e80349. [Google Scholar] [CrossRef] [PubMed]
- Okuno, Y.; Nakamura-Ishizu, A.; Otsu, K.; Suda, T.; Kubota, Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat. Med. 2012, 18, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Ohba, Y.; Akiyama, K.; Ohga, N.; Hamada, J.-I.; Nagao-Kitamoto, H.; Alam, M.T.; Yamamoto, K.; Kawamoto, T.; Inoue, N.; et al. Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Sci. Rep. 2016, 6, 28039. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Jain, R.K. Tumor microvasculature and microenvironment: Targets for anti- angiogenesis and normalization. Microvasc. Res. 2007, 74, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Anand, V.; Roy, S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J. Neuroimmune Pharmacol. 2014, 9, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular 02 tension (dioxin receptor/erythropoietin/hypoxia/transcription). Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, S.A.; de Andrade Júnior, D.R. HIF-1alpha and infectious diseases: A new frontier for the development of new therapies. Rev. Inst. Med. Trop. Sao Paulo 2017, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q. VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines 2018, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain: A hypoxic switch. Science (80-) 2002, 295, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef] [PubMed]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. J. Biol. Chem. 1997, 14, 3470–3481. [Google Scholar]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. BioMed Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Dachs, G.U.; Gleadle, J.M.; Nicholls, L.G.; Harris, A.L.; Stratford, I.J.; Hankinson, O.; Pugh, C.W.; Ratcliffe, P.J. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1997, 94, 8104–8109. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Bacon, A.; Harris, A. Hypoxia-inducible factors and hypoxic cell death in tumour physiology. Ann. Med. 2004, 36, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Kirito, K.; Hu, Y.; Komatsu, N. HIF-1 prevents the overproduction of mitochondrial ROS after cytokine stimulation through induction of PDK-1. Cell Cycle 2009, 8, 2844–2849. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ding, Z.; Hu, D.; Sun, F.; Dai, C.; Xie, J.; Hu, X. Central role of lactic acidosis in cancer cell resistance to glucose deprivation-induced cell death. J. Pathol. 2012, 227, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Mendler, A.N.; Hu, B.; Prinz, P.U.; Kreutz, M.; Gottfried, E.; Noessner, E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int. J. Cancer 2012, 131, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.R.; Eckhardt, B.L.; Cao, Y.; Pasqualini, R.; Argani, P.; Arap, W.; Ramsay, R.G.; Anderson, R.L. Inhibition of established micrometastases by targeted drug delivery via cell surface-associated GRP78. Clin. Cancer Res. 2013, 19, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.R.; Frudd, K.; Awad, W.; Hendershot, L.M. Endoplasmic Reticulum (ER) stress and Hypoxia response pathways interact to Potentiate Hypoxia-inducible Factor 1 (HIF-1) Transcriptional activity on targets like Vascular Endothelial Growth Factor (VEGF). J. Biol. Chem. 2014, 289, 3352–3364. [Google Scholar] [CrossRef] [PubMed]
- Giampietri, C.; Petrungaro, S.; Conti, S.; Facchiano, A.; Filippini, A.; Ziparo, E. Cancer Microenvironment and Endoplasmic Reticulum Stress Response. Mediators Inflamm. 2015, 2015, 417281. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. Physiological/pathological ramifications of transcription factors in the unfolded protein response. Genes Dev. 2017, 31, 1417–1438. [Google Scholar] [CrossRef] [PubMed]
- Karali, E.; Bellou, S.; Stellas, D.; Klinakis, A.; Murphy, C.; Fotsis, T. VEGF Signals through ATF6 and PERK to Promote Endothelial Cell Survival and Angiogenesis in the Absence of ER Stress. Mol. Cell 2014, 54, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour–stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015, 44–46, 94–112. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi, L.S.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-C.; Halford, M.M.; Achen, M.G.; Stacker, S.A. Exploring the role of endothelium in the tumour response to anti-angiogenic therapy. Biochem. Soc. Trans. 2014, 42, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Fontanella, C.; Ongaro, E.; Bolzonello, S.; Guardascione, M.; Fasola, G.; Aprile, G. Clinical advances in the development of novel VEGFR2 inhibitors. Ann. Transl. Med. 2014, 2, 123. [Google Scholar] [PubMed]
- Amadio, M.; Govoni, S.; Pascale, A. Targeting VEGF in eye neovascularization: What’s new?: A comprehensive review on current therapies and oligonucleotide-based interventions under development. Pharmacol. Res. 2016, 103, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Tewari, K.S.; Sill, M.W.; Long, H.J., III; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; et al. Improved Survival with Bevacizumab in Advanced Cervical Cancer. N. Engl. J. Med. 2014, 370, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Scartozzi, M.; Vincent, L.; Chiron, M.; Cascinu, S. Aflibercept, a New Way to Target Angiogenesis in the Second Line Treatment of Metastatic Colorectal Cancer (mCRC). Target. Oncol. 2016, 11, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E. Overall Survival in Renal-Cell Carcinoma with Pazopanib versus Sunitinib. N. Engl. J. Med. 2014, 370, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomised phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet (London, England) 2014, 384, 319–328. [Google Scholar] [CrossRef]
- van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.M.; Thijssen, V.L.; Griffioen, A.W. The Great Escape; the Hallmarks of Resistance to Antiangiogenic Therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Hoff, P.M.; Morris, J.S.; Wolff, R.A.; Eng, C.; Glover, K.Y.; Adinin, R.; Overman, M.J.; Valero, V.; Wen, S.; et al. Phase II Trial of Infusional Fluorouracil, Irinotecan, and Bevacizumab for Metastatic Colorectal Cancer: Efficacy and Circulating Angiogenic Biomarkers Associated With Therapeutic Resistance. J. Clin. Oncol. 2010, 28, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, N.S.; Reynolds, A.R. Anti-angiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Adjei, A.A. New Drug Development and Clinical Pharmacology Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist 2015, 20, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Petit, I.; Jin, D.; Rafii, S. The SDF-1-CXCR4 signaling pathway: A molecular hub modulating neo-angiogenesis. Trends Immunol. 2007, 28, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.L.; Gomes, M.P.; Catarino, R.J.; Rolfo, C.; Lopes, A.M.; Medeiros, R.M.; Araújo, A.M.; Coelho, A.L.; Gomes, M.P.; Catarino, R.J.; et al. Angiogenesis in NSCLC: Is vessel co-option the trunk that sustains the branches? Oncotarget 2017, 8, 39795–39804. [Google Scholar] [CrossRef] [PubMed]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.-H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.-N.; Tan, M.-H.; Yang, J.-P.; Cao, Y. Revisiting tumor angiogenesis: Vessel co-option, vessel remodeling, and cancer cell-derived vasculature formation. Chin. J. Cancer 2016, 35, 10. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Luo, H. Overview of advances in vasculogenic mimicry—A potential target for tumor therapy. Cancer Manag. Res. 2018, 10, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.K.; Seftor, E.A.; Fletcher, M.S.; Gardner, L.M.; Heidger, P.M.; Buller, R.E.; Seftor, R.E.; Hendrix, M.J. Molecular determinants of ovarian cancer plasticity. Am. J. Pathol. 2001, 158, 1279–1288. [Google Scholar] [CrossRef]
- Van Der Schaft, D.W.J.; Hillen, F.; Pauwels, P.; Kirschmann, D.A.; Castermans, K.; Egbrink, M.G.A.; Tran, M.G.B.; Sciot, R.; Hauben, E.; Hogendoorn, P.C.W.; et al. Tumor Cell Plasticity in Ewing Sarcoma, an Alternative Circulatory System Stimulated by Hypoxia. Cancer Res. 2005, 65, 11520–11529. [Google Scholar] [CrossRef] [PubMed]
- Hillen, F.; Baeten, C.I.M.; van de Winkel, A.; Creytens, D.; van der Schaft, D.W.J.; Winnepenninckx, V.; Griffioen, A.W. Leukocyte infiltration and tumor cell plasticity are parameters of aggressiveness in primary cutaneous melanoma. Cancer Immunol. Immunother. 2008, 57, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Reis e Sousa, C. Dendritic cells in a mature age. Nat. Rev. Immunol. 2006, 6, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar] [PubMed]
- Oyama, T.; Ran, S.; Ishida, T.; Nadaf, S.; Kerr, L.; Carbone, D.P.; Gabrilovich, D.I. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J. Immunol. 1998, 160, 1224–1232. [Google Scholar] [PubMed]
- Mimura, K.; Kono, K.; Takahashi, A.; Kawaguchi, Y.; Fujii, H. Vascular endothelial growth factor inhibits the function of human mature dendritic cells mediated by VEGF receptor-2. Cancer Immunol. Immunother. 2007, 56, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Kinashi, T. Intracellular signalling controlling integrin activation in lymphocytes. Nat. Rev. Immunol. 2005, 5, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Huang, S.J.; Murphy, G.F.; Mollet, I.G.; Hijnen, D.; Muthukuru, M.; Schanbacher, C.F.; Edwards, V.; Miller, D.M.; Kim, J.E.; et al. Human squamous cell carcinomas evade the immune response by down-regulation of vascular E-selectin and recruitment of regulatory T cells. J. Exp. Med. 2008, 205, 2221–2234. [Google Scholar] [CrossRef] [PubMed]
- Afanasiev, O.K.; Nagase, K.; Simonson, W.; Vandeven, N.; Koelle, D.M.; Clark, R.; Nghiem, P. Vascular E-selectin expression correlates with CD8 lymphocyte infiltration and improved outcome in Merkel cell carcinoma. J. Investig. Dermatol. 2013, 133, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Damen, C.A.; Martinotti, S.; Blijham, G.H.; Groenewegen, G. Endothelial Intercellular Adhesion Molecule-i Expression Is Suppressed in Human. Cancer Res. 1996, 1, 1111–1117. [Google Scholar]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lalani, A.S.; Harding, T.C.; Luan, B.; Koprivnikar, K.; Huan Tu, G.; Prell, R.; VanRoey, M.J.; Simmons, A.D.; Jooss, K. Vascular Endothelial Growth Factor Blockade Reduces Intratumoral Regulatory T Cells and Enhances the Efficacy of a GM-CSF-Secreting Cancer Immunotherapy. Clin. Cancer Res. 2006, 12, 6808–6816. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Suzuki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. 2009, 29, 881–888. [Google Scholar] [PubMed]
- Alfaro, C.; Suarez, N.; Gonzalez, A.; Solano, S.; Erro, L.; Dubrot, J.; Palazon, A.; Hervas-Stubbs, S.; Gurpide, A.; Lopez-Picazo, J.M.; et al. Influence of bevacizumab, sunitinib and sorafenib as single agents or in combination on the inhibitory effects of VEGF on human dendritic cell differentiation from monocytes. Br. J. Cancer 2009, 100, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Borgström, P.; Hughes, G.K.; Hansell, P.; Wolitsky, B.A.; Sriramarao, P. Leukocyte adhesion in angiogenic blood vessels. Role of E-selectin, P-selectin, and beta2 integrin in lymphotoxin-mediated leukocyte recruitment in tumor microvessels. J. Clin. Investig. 1997, 99, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Eruslanov, E.; Kübler, H.; Tseng, T.; Sakai, Y.; Su, Z.; Kaliberov, S.; Heiser, A.; Rosser, C.; Dahm, P.; et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: Link to tumor-induced immune suppression in renal cell carcinoma. J. Immunol. 2008, 181, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Adotevi, O.; Pere, H.; Ravel, P.; Haicheur, N.; Badoual, C.; Merillon, N.; Medioni, J.; Peyrard, S.; Roncelin, S.; Verkarre, V.; et al. A Decrease of Regulatory T Cells Correlates With Overall Survival After Sunitinib-based Antiangiogenic Therapy in Metastatic Renal Cancer Patients. J. Immunother. 2010, 33, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Gao, Z.; Li, X.; Dong, L.; Han, W.; Nie, J. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PDL1 blockade. Oncotarget 2017, 8, 110693–110707. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-D.; Hoang, A. Resistance to anti-angiogenic therapy is associated with an immunosuppressive tumor microenvironment in metastatic renal cell carcinoma. Cancer Immunol. Res. 2015, 3, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Guislain, A.; Gadiot, J.; Kaiser, A.; Jordanova, E.S.; Broeks, A.; Sanders, J.; van Boven, H.; de Gruijl, T.D.; Haanen, J.B.A.G.; Bex, A.; et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol. Immunother. 2015, 64, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.-L.; et al. IMmotion151: A Randomized Phase III Study of Atezolizumab Plus Bevacizumab vs. Sunitinib in Untreated Metastatic Renal Cell Carcinoma (mRCC). J. Clin. Oncol. 2018, 36, 578. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Activators | Functions | Inhibitors | Functions |
|---|---|---|---|
| Vascular endothelial growth factor family | Induction of angiogenesis, enhancement of vascular permeability | Angiopoietin-2 | Antagonist of Ang1 |
| Epidermal growth factor | Promotes growth of vascular endothelial cells | Thrombospondin-1,2 | Inhibits endothelial migration, growth, adhesion and survival |
| Fibroblast growth factor | Induction of angiogenesis | collagen | Substrate for MMPs |
| Platelet-derived growth factor | Involved in migration of vascular endothelial cells | Endostatin | Inhibits endothelial survival and migration |
| Angiopoietin-1 | Stabilization of vascular endothelium | Angiostatin | Suppresses tumor angiogenesis |
| Transforming growth factor | Production of extracellular matrix | TIMPs | Suppresses pathological angiogenesis |
| Ephrin | Control of blood vessel and lymph duct formation | Platelet Factor-4 | Inhibits binding of bFGF and VEGF |
| Matrix metalloproteinase | Degradation of extracellular matrix, activation of angiogenesis inducing factor | Vasostatin | Inhibits endothelial growth |
| Drug | Target Molecule | Approved Disease |
|---|---|---|
| Bevacizumab | Anti-VEGF monoclonal antibody | mCRC, NSCLC, mRCC, ovarian cancer, malignant glioma, advanced cervical cancer, fallopian tube cancer, primary peritoneal cancer |
| Ramucirumab | Anti-VEGFR2 monoclonal antibody | Advanced gastric or gastroesophageal junction adenocarcinoma, NSCLC, advanced colorectal cancer |
| Ziv-aflibercept | Soluble decoy of VEGFR | Metastatic colorectal cancer |
| Sunitinib | TKI: VEGFR, PDGFR, FLT3, KIT | RCC, Gastrointestinal stromal tumor, pancreatic neuroendocrine tumor |
| Sorafenib | TKI: VEGFR, PDGFR, FLT3, KIT, Raf | RCC, unresectable hepatocellular carcinoma, metastatic or recurrent thyroid carcinoma |
| Axitinib | TKI: VEGFR, PDGFR, KIT | Advanced RCC |
| Pazopanib | Multiple targeted receptor TKI | RCC, Advanced soft tissue sarcoma |
| Vandetanib | TKI: VEGFR, EGFR, RET | Unresectable or metastatic medullary thyroid cancer |
| Tumor Type | Combination Drugs | Study Status | NCT ID |
|---|---|---|---|
| Stage IV NSCLC | Atezolizumab+Carboplatin+paclitaxel+Bevacizumab | Active, not recruiting | NCT02366143 |
| Advanced RCC | Bevacizumab+Atezolizumab | Active, not recruiting | NCT02420821 |
| Advanced RCC | Avelumab+Axitinib | Active, not recruiting | NCT02684006 |
| Advanced RCC | Lenvatinib/Everolimus or Lenvatinib/Pembrolizumab | Recruiting | NCT02811861 |
| Recurrent OC, FTC, or PPC | Pegylated Liposomal Doxorubicin+Atezolizumab+Bevacizumab | Recruiting | NCT02839707 |
| RCC | Pembrolizumab+Axitinib | Active, not recruiting | NCT02853331 |
| Late relapse OC | Atezolizumab+Chemotherapy+Bevacizumab | Recruiting | NCT02891824 |
| OC,FTC,or PPC | Atezolizumab+Carboplatin+paclitaxel+Bevacizumab | Recruiting | NCT03038100 |
| Early relapse OC | Atezolizumab+Bevacizumab+Chemotherapy | Recruiting | NCT03353831 |
| Locally Advanced or Metasatatic HCC | Atezolizumab+Bevacizumab | Recruiting | NCT03434379 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katayama, Y.; Uchino, J.; Chihara, Y.; Tamiya, N.; Kaneko, Y.; Yamada, T.; Takayama, K. Tumor Neovascularization and Developments in Therapeutics. Cancers 2019, 11, 316. https://doi.org/10.3390/cancers11030316
Katayama Y, Uchino J, Chihara Y, Tamiya N, Kaneko Y, Yamada T, Takayama K. Tumor Neovascularization and Developments in Therapeutics. Cancers. 2019; 11(3):316. https://doi.org/10.3390/cancers11030316
Chicago/Turabian StyleKatayama, Yuki, Junji Uchino, Yusuke Chihara, Nobuyo Tamiya, Yoshiko Kaneko, Tadaaki Yamada, and Koichi Takayama. 2019. "Tumor Neovascularization and Developments in Therapeutics" Cancers 11, no. 3: 316. https://doi.org/10.3390/cancers11030316
APA StyleKatayama, Y., Uchino, J., Chihara, Y., Tamiya, N., Kaneko, Y., Yamada, T., & Takayama, K. (2019). Tumor Neovascularization and Developments in Therapeutics. Cancers, 11(3), 316. https://doi.org/10.3390/cancers11030316