Oncogenic Deregulation of Cell Adhesion Molecules in Leukemia


 and
and
Abstract
1. Introduction
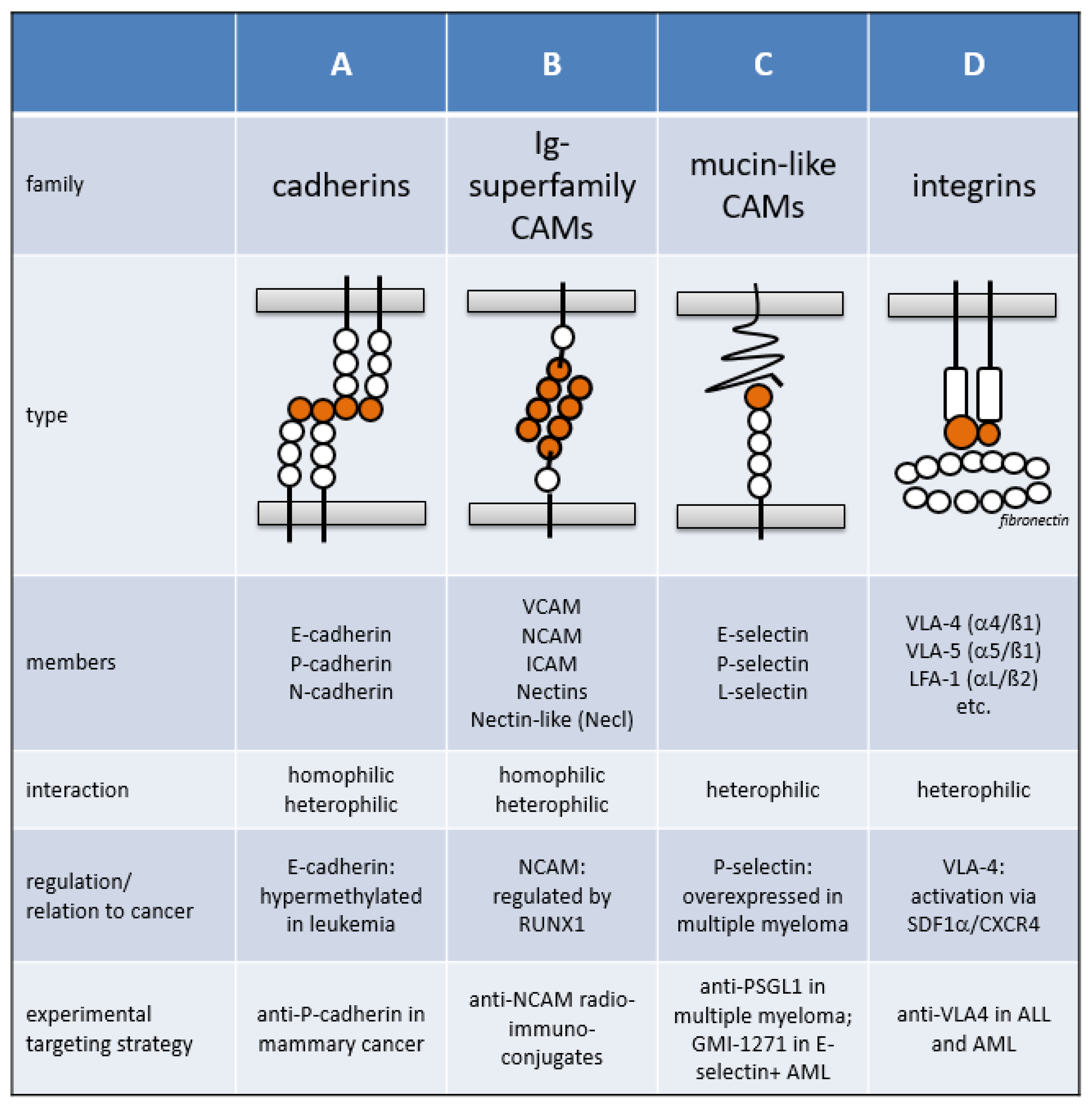
1.1. Cell Adhesion Molecule Families
1.1.1. The Cadherin Superfamily
1.1.2. The Immunoglobulin Superfamily
1.1.3. Mucin-like CAMs
1.1.4. The Integrin Family
1.2. Adhesion Molecules of the Hematopoietic Stem Cell Niche
1.3. Oncogenic Lesions in Myeloid Leukemia
2. Oncogenes Directly Deregulate Cellular Adhesion
3. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Prakasam, A.K.; Maruthamuthu, V.; Leckband, D.E. Similarities between heterophilic and homophilic cadherin adhesion. Proc. Natl. Acad. Sci. USA 2006, 103, 15434–15439. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F. Beyond E-cadherin: Roles of other cadherin superfamily members in cancer. Nat. Rev. Cancer 2014, 14, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.C.; Yan, Z.; Zhang, Q.; Kuszpit, K.; Zasadny, K.; Qiu, M.; Painter, C.L.; Wong, A.; Kraynov, E.; Arango, M.E.; et al. PF-03732010: A fully human monoclonal antibody against P-cadherin with antitumor and antimetastatic activity. Clin. Cancer Res. 2010, 16, 5177–5188. [Google Scholar] [CrossRef] [PubMed]
- Aggerholm, A.; Holm, M.S.; Guldberg, P.; Olesen, L.H.; Hokland, P. Promoter hypermethylation of p15INK4B, HIC1, CDH1, and ER is frequent in myelodysplastic syndrome and predicts poor prognosis in early-stage patients. Eur. J. Haematol. 2006, 76, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Melki, J.R.; Vincent, P.C.; Brown, R.D.; Clark, S.J. Hypermethylation of E-cadherin in leukemia. Blood 2000, 95, 3208–3213. [Google Scholar] [PubMed]
- Rao, Y.; Wang, H.; Fan, L.; Chen, G. Silencing MTA1 by RNAi reverses adhesion, migration and invasiveness of cervical cancer cells (SiHa) via altered expression of p53, and E-cadherin/beta-catenin complex. J. Huazhong Univ. Sci. Technol. Med. Sci. 2011, 31, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Chisholm, K.M.; Ma, L.; Arber, D.A. E-cadherin is a specific marker for erythroid differentiation and has utility, in combination with CD117 and CD34, for enumerating myeloblasts in hematopoietic neoplasms. Am. J. Clin. Pathol. 2014, 141, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Arai, F.; Hosokawa, K.; Toyama, H.; Matsumoto, Y.; Suda, T. Role of N-cadherin in the regulation of hematopoietic stem cells in the bone marrow niche. Ann. N. Y. Acad. Sci. 2012, 1266, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Zhi, L.; Gao, Y.; Yu, C.; Zhang, Y.; Zhang, B.; Yang, J.; Yao, Z. N-Cadherin Aided in Maintaining the Characteristics of Leukemic Stem Cells. Anat. Rec. (Hoboken.) 2016, 299, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Dessein, P.H.; Joffe, B.I.; Singh, S. Biomarkers of endothelial dysfunction, cardiovascular risk factors and atherosclerosis in rheumatoid arthritis. Arthritis Res. 2005, 7, R634–R643. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Zhang, S.Y.; Chen, Y.Y.; Shi, K.; Zou, B.; Liu, J.; Yang, Q.; Jiang, H.; Wei, L.; Li, C.Z.; et al. ICAM-1 Deficiency in the Bone Marrow Niche Impairs Quiescence and Repopulation of Hematopoietic Stem Cells. Stem Cell Rep. 2018, 11, 258–273. [Google Scholar]
- Conrad, F.; Zhu, X.; Zhang, X.; Chalkley, R.J.; Burlingame, A.L.; Marks, J.D.; Liu, B. Human antibodies targeting cell surface antigens overexpressed by the hormone refractory metastatic prostate cancer cells: ICAM-1 is a tumor antigen that mediates prostate cancer cell invasion. J. Mol. Med. (Berl.) 2009, 87, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Huang, J.; Wang, L.; Jia, D.; Yang, J.; Dillon, D.A.; Zurakowski, D.; Mao, H.; Moses, M.A.; Auguste, D.T. ICAM-1 as a molecular target for triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14710–14715. [Google Scholar] [CrossRef] [PubMed]
- Klausz, K.; Cieker, M.; Kellner, C.; Oberg, H.H.; Kabelitz, D.; Valerius, T.; Burger, R.; Gramatzki, M.; Peipp, M. A novel Fc-engineered human ICAM-1/CD54 antibody with potent anti-myeloma activity developed by cellular panning of phage display libraries. Oncotarget 2017, 8, 77552–77566. [Google Scholar] [CrossRef] [PubMed]
- Veitonmaki, N.; Hansson, M.; Zhan, F.; Sundberg, A.; Lofstedt, T.; Ljungars, A.; Li, Z.C.; Martinsson-Niskanen, T.; Zeng, M.; Yang, Y.; et al. A human ICAM-1 antibody isolated by a function-first approach has potent macrophage-dependent antimyeloma activity in vivo. Cancer Cell 2013, 23, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Wichert, S.; Juliusson, G.; Johansson, A.; Sonesson, E.; Teige, I.; Wickenberg, A.T.; Frendeus, B.; Korsgren, M.; Hansson, M. A single-arm, open-label, phase 2 clinical trial evaluating disease response following treatment with BI-505, a human anti-intercellular adhesion molecule-1 monoclonal antibody, in patients with smoldering multiple myeloma. PLoS ONE 2017, 12, e0171205. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.M.; Berezin, V.; Bock, E. Signaling mechanisms of neurite outgrowth induced by the cell adhesion molecules NCAM and N-cadherin. Cell. Mol. Life Sci. 2008, 65, 3809–3821. [Google Scholar] [CrossRef] [PubMed]
- Satoh-Horikawa, K.; Nakanishi, H.; Takahashi, K.; Miyahara, M.; Nishimura, M.; Tachibana, K.; Mizoguchi, A.; Takai, Y. Nectin-3, a new member of immunoglobulin-like cell adhesion molecules that shows homophilic and heterophilic cell-cell adhesion activities. J. Biol. Chem. 2000, 275, 10291–10299. [Google Scholar] [CrossRef] [PubMed]
- Kurita, S.; Ogita, H.; Takai, Y. Cooperative role of nectin-nectin and nectin-afadin interactions in formation of nectin-based cell-cell adhesion. J. Biol. Chem. 2011, 286, 36297–36303. [Google Scholar] [CrossRef] [PubMed]
- Takai, Y.; Nakanishi, H. Nectin and afadin: Novel organizers of intercellular junctions. J. Cell Sci. 2003, 116, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Rikitake, Y.; Takai, Y. Directional cell migration regulation by small G proteins, nectin-like molecule-5, and afadin. Int. Rev. Cell Mol. Biol. 2011, 287, 97–143. [Google Scholar] [PubMed]
- Blake, S.J.; Dougall, W.C.; Miles, J.J.; Teng, M.W.; Smyth, M.J. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 5183–5188. [Google Scholar] [CrossRef] [PubMed]
- Stamm, H.; Wellbrock, J.; Fiedler, W. Interaction of PVR/PVRL2 with TIGIT/DNAM-1 as a novel immune checkpoint axis and therapeutic target in cancer. Mamm. Genome 2018, 29, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012, 2012, 676731. [Google Scholar] [CrossRef] [PubMed]
- Kansas, G.S. Selectins and their ligands: Current concepts and controversies. Blood 1996, 88, 3259–3287. [Google Scholar] [PubMed]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.M.; Corbel, S.Y.; Merzaban, J.S.; Carlow, D.A.; Gossens, K.; Duenas, J.; So, L.; Yi, L.; Ziltener, H.J. Recruitment of adult thymic progenitors is regulated by P-selectin and its ligand PSGL-1. Nat. Immunol. 2005, 6, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Frenette, P.S.; Denis, C.V.; Weiss, L.; Jurk, K.; Subbarao, S.; Kehrel, B.; Hartwig, J.H.; Vestweber, D.; Wagner, D.D. P-Selectin glycoprotein ligand 1 (PSGL-1) is expressed on platelets and can mediate platelet-endothelial interactions in vivo. J. Exp. Med. 2000, 191, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Veerman, K.M.; Williams, M.J.; Uchimura, K.; Singer, M.S.; Merzaban, J.S.; Naus, S.; Carlow, D.A.; Owen, P.; Rivera-Nieves, J.; Rosen, S.D.; et al. Interaction of the selectin ligand PSGL-1 with chemokines CCL21 and CCL19 facilitates efficient homing of T cells to secondary lymphoid organs. Nat. Immunol. 2007, 8, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. The role of selectins in inflammation and disease. Trends Mol. Med. 2003, 9, 263–268. [Google Scholar] [CrossRef]
- Barthel, S.R.; Gavino, J.D.; Descheny, L.; Dimitroff, C.J. Targeting selectins and selectin ligands in inflammation and cancer. Expert Opin. Targets 2007, 11, 1473–1491. [Google Scholar] [CrossRef] [PubMed]
- Sipkins, D.A.; Wei, X.; Wu, J.W.; Runnels, J.M.; Cote, D.; Means, T.K.; Luster, A.D.; Scadden, D.T.; Lin, C.P. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005, 435, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Erbani, J.M.; Barbier, V.; Davies, J.M.; Tay, J.; Fiveash, C.E.; Lowe, J.; Tallack, M.; Magnani, J.L.; Levesque, J.-P. Vascular E-Selectin Mediates Chemo-Resistance in Acute Myeloid Leukemia Initiating Cells Via Canonical Receptors PSGL-1 (CD162) and Hcell (CD44) and AKT Signaling. Blood 2017, 130, 793. [Google Scholar]
- Kim, K.J.; Kwon, S.H.; Yun, J.H.; Jeong, H.S.; Kim, H.R.; Lee, E.H.; Ye, S.K.; Cho, C.H. STAT3 activation in endothelial cells is important for tumor metastasis via increased cell adhesion molecule expression. Oncogene 2017, 36, 5445–5459. [Google Scholar] [CrossRef] [PubMed]
- Hauselmann, I.; Roblek, M.; Protsyuk, D.; Huck, V.; Knopfova, L.; Grassle, S.; Bauer, A.T.; Schneider, S.W.; Borsig, L. Monocyte Induction of E-Selectin-Mediated Endothelial Activation Releases VE-Cadherin Junctions to Promote Tumor Cell Extravasation in the Metastasis Cascade. Cancer Res. 2016, 76, 5302–5312. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Bendall, L.J.; Bradstock, K.F. G-CSF: From granulopoietic stimulant to bone marrow stem cell mobilizing agent. Cytokine Growth Factor Rev. 2014, 25, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Wallstabe, L.; Mades, A.; Frenz, S.; Einsele, H.; Rader, C.; Hudecek, M. CAR T cells targeting alphavbeta3 integrin are effective against advanced cancer in preclinical models. Adv. Cell Gene 2018, 1, e11. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Trumpp, A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 2011, 208, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, C.; Asada, N.; Frenette, P.S. The hematopoietic stem cell niche: From embryo to adult. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Lo Celso, C.; Scadden, D.T. The haematopoietic stem cell niche at a glance. J. Cell Sci. 2011, 124, 3529–3535. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Stier, S.; Ko, Y.; Forkert, R.; Lutz, C.; Neuhaus, T.; Grunewald, E.; Cheng, T.; Dombkowski, D.; Calvi, L.M.; Rittling, S.R.; et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J. Exp. Med. 2005, 201, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Tabone-Eglinger, S.; Calderin-Sollet, Z.; Pinon, P.; Aebischer, N.; Wehrle-Haller, M.; Jacquier, M.C.; Boettiger, D.; Wehrle-Haller, B. Niche anchorage and signaling through membrane-bound Kit-ligand/c-kit receptor are kinase independent and imatinib insensitive. FASEB J. 2014, 28, 4441–4456. [Google Scholar] [CrossRef] [PubMed]
- Dimitroff, C.J.; Lee, J.Y.; Rafii, S.; Fuhlbrigge, R.C.; Sackstein, R. CD44 is a major E-selectin ligand on human hematopoietic progenitor cells. J. Cell Biol. 2001, 153, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Godavarthy, P.S.; Herkt, S.; Hayduk, N.; Weissenberger, E.; Manavski, Y.; Lucas, T.; Pan, K.-T.; Voutsinas, J.M.; Wu, Q.V.; Mueller, M.C. The Vascular Bone Marrow Niche Influences Outcome in Chronic Myeloid Leukemia. Blood 2018, 132, 3846. [Google Scholar]
- Craddock, C.F.; Nakamoto, B.; Andrews, R.G.; Priestley, G.V.; Papayannopoulou, T. Antibodies to VLA4 integrin mobilize long-term repopulating cells and augment cytokine-induced mobilization in primates and mice. Blood 1997, 90, 4779–4788. [Google Scholar] [PubMed]
- Papayannopoulou, T.; Nakamoto, B. Peripheralization of hemopoietic progenitors in primates treated with anti-VLA4 integrin. Proc. Natl. Acad. Sci. USA 1993, 90, 9374–9378. [Google Scholar] [CrossRef] [PubMed]
- Rettig, M.P.; Ansstas, G.; DiPersio, J.F. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia 2012, 26, 34–53. [Google Scholar] [CrossRef] [PubMed]
- Shishido, S.; Bonig, H.; Kim, Y.M. Role of integrin alpha4 in drug resistance of leukemia. Front. Oncol. 2014, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Van der Loo, J.C.; Xiao, X.; McMillin, D.; Hashino, K.; Kato, I.; Williams, D.A. VLA-5 is expressed by mouse and human long-term repopulating hematopoietic cells and mediates adhesion to extracellular matrix protein fibronectin. J. Clin. Investig. 1998, 102, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Motabi, I.H.; DiPersio, J.F. Advances in stem cell mobilization. Blood Rev. 2012, 26, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Cutler, C.; Antin, J.H. Peripheral blood stem cells for allogeneic transplantation: A review. Stem Cells 2001, 19, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Giralt, S.; Costa, L.; Schriber, J.; Dipersio, J.; Maziarz, R.; McCarty, J.; Shaughnessy, P.; Snyder, E.; Bensinger, W.; Copelan, E.; et al. Optimizing autologous stem cell mobilization strategies to improve patient outcomes: Consensus guidelines and recommendations. Biol Blood Marrow Transpl. 2014, 20, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013, 19, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Gilliland, D.G. Genetics of myeloid leukemias. Annu. Rev. Genom. Hum. Genet. 2002, 3, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Saglio, G. Molecular pathways: BCR-ABL. Clin. Cancer Res. 2012, 18, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Lazarides, K.; Lewis, J.B.; von Andrian, U.H.; Van Etten, R.A. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 2014, 123, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Lazarides, K.; von Andrian, U.H.; Van Etten, R.A. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat. Med. 2006, 12, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Carlesso, N.; Griffin, J.D.; Hemler, M.E. Bcr/Abl expression stimulates integrin function in hematopoietic cell lines. J. Clin. Investig. 1996, 98, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Guller, S.; Beissert, T.; Puccetti, E.; Ruthardt, M. BCR and its mutants, the reciprocal t(9;22)-associated ABL/BCR fusion proteins, differentially regulate the cytoskeleton and cell motility. BMC Cancer 2006, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.K.; Cancelas, J.A.; Zheng, Y.; Williams, D.A. Rac GTPases as key regulators of p210-BCR-ABL-dependent leukemogenesis. Leukemia 2008, 22, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.; Small, D. FLT3: ITDoes matter in leukemia. Leukemia 2003, 17, 1738–1752. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.Y.; Man, C.H.; Kwong, Y.L. FLT3 inhibition: A moving and evolving target in acute myeloid leukaemia. Leukemia 2013, 27, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Katsumi, A.; Kiyoi, H.; Abe, A.; Tanizaki, R.; Iwasaki, T.; Kobayashi, M.; Matsushita, T.; Kaibuchi, K.; Senga, T.; Kojima, T.; et al. FLT3/ ITD regulates leukaemia cell adhesion through alpha4beta1 integrin and Pyk2 signalling. Eur. J. Haematol. 2011, 86, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Solanilla, A.; Grosset, C.; Duchez, P.; Legembre, P.; Pitard, V.; Dupouy, M.; Belloc, F.; Viallard, J.F.; Reiffers, J.; Boiron, J.M.; et al. Flt3-ligand induces adhesion of haematopoietic progenitor cells via a very late antigen (VLA)-4- and VLA-5-dependent mechanism. Br. J. Haematol. 2003, 120, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, A.; Wolach, O.; El-Jawahri, A.; Ho, V.T.; DiPersio, J.F.; Soiffer, R.; Stone, R.M.; Chen, Y.B. The effect of FLT3-ITD and NPM1 mutation on survival in intensively treated elderly patients with cytogenetically normal acute myeloid leukemia. Leuk. Lymphoma 2016, 57, 1977–1979. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Zeng, D.; Shen, Z.; Liao, J.; Wang, X.; Liu, Y.; Zhang, X.; Kong, P. Integrin alphavbeta3 enhances beta-catenin signaling in acute myeloid leukemia harboring Fms-like tyrosine kinase-3 internal tandem duplication mutations: Implications for microenvironment influence on sorafenib sensitivity. Oncotarget 2016, 7, 40387–40397. [Google Scholar] [CrossRef] [PubMed]
- Reiter, K.; Polzer, H.; Krupka, C.; Maiser, A.; Vick, B.; Rothenberg-Thurley, M.; Metzeler, K.H.; Dorfel, D.; Salih, H.R.; Jung, G.; et al. Tyrosine kinase inhibition increases the cell surface localization of FLT3-ITD and enhances FLT3-directed immunotherapy of acute myeloid leukemia. Leukemia 2018, 32, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.G.; Al-Shahrour, F.; Hartwell, K.A.; Chu, L.P.; Jaras, M.; Puram, R.V.; Puissant, A.; Callahan, K.P.; Ashton, J.; McConkey, M.E.; et al. In Vivo RNAi screening identifies a leukemia-specific dependence on integrin beta 3 signaling. Cancer Cell 2013, 24, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Passamonti, F.; Rambaldi, A.; Barosi, G.; Rosen, P.J.; Rumi, E.; Gattoni, E.; Pieri, L.; Guglielmelli, P.; Elena, C.; et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014, 120, 513–520. [Google Scholar] [CrossRef] [PubMed]
- De Grandis, M.; Cambot, M.; Wautier, M.P.; Cassinat, B.; Chomienne, C.; Colin, Y.; Wautier, J.L.; Le Van Kim, C.; El Nemer, W. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood 2013, 121, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; El Nemer, W.; Gane, P.; Rain, J.D.; Cartron, J.P.; Colin, Y.; Le Van Kim, C.; Wautier, J.L. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin alpha5 chain and Lu/BCAM. Blood 2007, 110, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.I. Molecular basis of the diagnosis and treatment of polycythemia vera and essential thrombocythemia. Blood 2006, 107, 4214–4222. [Google Scholar] [CrossRef] [PubMed]
- El Nemer, W.; Colin, Y.; Le Van Kim, C. Role of Lu/BCAM glycoproteins in red cell diseases. Transfus. Clin. Biol. 2010, 17, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Pabst, T.; Mueller, B.U.; Zhang, P.; Radomska, H.S.; Narravula, S.; Schnittger, S.; Behre, G.; Hiddemann, W.; Tenen, D.G. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat. Genet. 2001, 27, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.Y.; Hou, H.A.; Chen, Y.K.; Li, L.Y.; Chen, P.H.; Tseng, M.H.; Huang, C.F.; Lee, F.Y.; Liu, M.C.; Liu, C.W.; et al. The N-terminal CEBPA mutant in acute myeloid leukemia impairs CXCR4 expression. Haematologica 2014, 99, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Tavor, S.; Petit, I.; Porozov, S.; Avigdor, A.; Dar, A.; Leider-Trejo, L.; Shemtov, N.; Deutsch, V.; Naparstek, E.; Nagler, A.; et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004, 64, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Spoo, A.C.; Lubbert, M.; Wierda, W.G.; Burger, J.A. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood 2007, 109, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Dufour, A.; Schneider, F.; Metzeler, K.H.; Hoster, E.; Schneider, S.; Zellmeier, E.; Benthaus, T.; Sauerland, M.C.; Berdel, W.E.; Buchner, T.; et al. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J. Clin. Oncol. 2010, 28, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Preudhomme, C.; Sagot, C.; Boissel, N.; Cayuela, J.M.; Tigaud, I.; de Botton, S.; Thomas, X.; Raffoux, E.; Lamandin, C.; Castaigne, S.; et al. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: A study from the Acute Leukemia French Association (ALFA). Blood 2002, 100, 2717–2723. [Google Scholar] [CrossRef] [PubMed]
- Gattenloehner, S.; Chuvpilo, S.; Langebrake, C.; Reinhardt, D.; Muller-Hermelink, H.K.; Serfling, E.; Vincent, A.; Marx, A. Novel RUNX1 isoforms determine the fate of acute myeloid leukemia cells by controlling CD56 expression. Blood 2007, 110, 2027–2033. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Dicker, F.; Kern, W.; Wendland, N.; Sundermann, J.; Alpermann, T.; Haferlach, C.; Haferlach, T. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood 2011, 117, 2348–2357. [Google Scholar] [CrossRef] [PubMed]
- Raspadori, D.; Damiani, D.; Lenoci, M.; Rondelli, D.; Testoni, N.; Nardi, G.; Sestigiani, C.; Mariotti, C.; Birtolo, S.; Tozzi, M.; et al. CD56 antigenic expression in acute myeloid leukemia identifies patients with poor clinical prognosis. Leukemia 2001, 15, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Sasca, D.; Schueler, A.; Szybinski, J.; Kriege, O.; Kunz, K.; Fehr, E.M.; Haehnel, P.; Gebhardt, W.H.; Reid, G.; Theobald, M. Targeting Aberrant Ncam (neural cell adhesion molecule; CD56) Expression in Acute Myeloid Leukemia: Am Soc Hematology. Blood 2015, 126, 311. [Google Scholar]
- Bourgeois, M.; Bailly, C.; Frindel, M.; Guerard, F.; Cherel, M.; Faivre-Chauvet, A.; Kraeber-Bodere, F.; Bodet-Milin, C. Radioimmunoconjugates for treating cancer: Recent advances and current opportunities. Expert Opin. Biol. 2017, 17, 813–819. [Google Scholar]
- Jensen, M.; Berthold, F. Targeting the neural cell adhesion molecule in cancer. Cancer Lett. 2007, 258, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, K.; Chen-Wichmann, L.; Kuvardina, O.N.; Lausen, J.; Henschler, R.; Wichmann, C. The truncated RUNX1/ETO activates VLA-4-dependent adhesion and migration of hematopoietic progenitor cells. Haematologica 2014, 99, e253–e256. [Google Scholar] [CrossRef] [PubMed]
- Ptasinska, A.; Assi, S.A.; Martinez-Soria, N.; Imperato, M.R.; Piper, J.; Cauchy, P.; Pickin, A.; James, S.R.; Hoogenkamp, M.; Williamson, D.; et al. Identification of a dynamic core transcriptional network in t(8;21) AML that regulates differentiation block and self-renewal. Cell Rep. 2014, 8, 1974–1988. [Google Scholar] [CrossRef] [PubMed]
- Shia, W.J.; Okumura, A.J.; Yan, M.; Sarkeshik, A.; Lo, M.C.; Matsuura, S.; Komeno, Y.; Zhao, X.; Nimer, S.D.; Yates, J.R., 3rd; et al. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood 2012, 119, 4953–4962. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gural, A.; Sun, X.J.; Zhao, X.; Perna, F.; Huang, G.; Hatlen, M.A.; Vu, L.; Liu, F.; Xu, H.; et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 2011, 333, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.F.; Wang, Y.; Lo, M.C.; Yan, M.; Kanbe, E.; Zhang, D.E. The multi-functional cellular adhesion molecule CD44 is regulated by the 8;21 chromosomal translocation. Leukemia 2007, 21, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Puig-Kroger, A.; Sanchez-Elsner, T.; Ruiz, N.; Andreu, E.J.; Prosper, F.; Jensen, U.B.; Gil, J.; Erickson, P.; Drabkin, H.; Groner, Y.; et al. RUNX/AML and C/EBP factors regulate CD11a integrin expression in myeloid cells through overlapping regulatory elements. Blood 2003, 102, 3252–3261. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Liu, Y.; Lukasik, S.M.; Speck, N.A.; Bushweller, J.H. CBFbeta allosterically regulates the Runx1 Runt domain via a dynamic conformational equilibrium. Nat. Struct. Mol. Biol. 2004, 11, 901–906. [Google Scholar] [PubMed]
- Ponnusamy, K.; Kohrs, N.; Ptasinska, A.; Assi, S.A.; Herold, T.; Hiddemann, W.; Lausen, J.; Bonifer, C.; Henschler, R.; Wichmann, C. RUNX1/ETO blocks selectin-mediated adhesion via epigenetic silencing of PSGL-1. Oncogenesis 2015, 4, e146. [Google Scholar] [CrossRef] [PubMed]
- Saia, M.; Termanini, A.; Rizzi, N.; Mazza, M.; Barbieri, E.; Valli, D.; Ciana, P.; Gruszka, A.M.; Alcalay, M. AML1/ETO accelerates cell migration and impairs cell-to-cell adhesion and homing of hematopoietic stem/progenitor cells. Sci. Rep. 2016, 6, 34957. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Hofmann, J.; Burmeister, T.; Groger, D.; Park, T.S.; Emerenciano, M.; Pombo de Oliveira, M.; Renneville, A.; Villarese, P.; Macintyre, E.; et al. The MLL recombinome of acute leukemias in 2013. Leukemia 2013, 27, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Poirel, H.; Rack, K.; Delabesse, E.; Radford-Weiss, I.; Troussard, X.; Debert, C.; Leboeuf, D.; Bastard, C.; Picard, F.; Veil-Buzyn, A.; et al. Incidence and characterization of MLL gene (11q23) rearrangements in acute myeloid leukemia M1 and M5. Blood 1996, 87, 2496–2505. [Google Scholar] [PubMed]
- Su, L.; Hattori, M.; Moriyama, M.; Murata, N.; Harazaki, M.; Kaibuchi, K.; Minato, N. AF-6 controls integrin-mediated cell adhesion by regulating Rap1 activation through the specific recruitment of Rap1GTP and SPA-1. J. Biol. Chem. 2003, 278, 15232–15238. [Google Scholar] [CrossRef] [PubMed]
- Manara, E.; Baron, E.; Tregnago, C.; Aveic, S.; Bisio, V.; Bresolin, S.; Masetti, R.; Locatelli, F.; Basso, G.; Pigazzi, M. MLL-AF6 fusion oncogene sequesters AF6 into the nucleus to trigger RAS activation in myeloid leukemia. Blood 2014, 124, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Fournier, G.; Cabaud, O.; Josselin, E.; Chaix, A.; Adelaide, J.; Isnardon, D.; Restouin, A.; Castellano, R.; Dubreuil, P.; Chaffanet, M.; et al. Loss of AF6/afadin, a marker of poor outcome in breast cancer, induces cell migration, invasiveness and tumor growth. Oncogene 2011, 30, 3862–3874. [Google Scholar] [CrossRef] [PubMed]
- Nuchprayoon, I.; Meyers, S.; Scott, L.M.; Suzow, J.; Hiebert, S.; Friedman, A.D. PEBP2/CBF, the murine homolog of the human myeloid AML1 and PEBP2 beta/CBF beta proto-oncoproteins, regulates the murine myeloperoxidase and neutrophil elastase genes in immature myeloid cells. Mol. Cell. Biol. 1994, 14, 5558–5568. [Google Scholar] [PubMed]
- Azab, A.K.; Quang, P.; Azab, F.; Pitsillides, C.; Thompson, B.; Chonghaile, T.; Patton, J.T.; Maiso, P.; Monrose, V.; Sacco, A.; et al. P-selectin glycoprotein ligand regulates the interaction of multiple myeloma cells with the bone marrow microenvironment. Blood 2012, 119, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- De la Puente, P.; Azab, A.K. Contemporary drug therapies for multiple myeloma. Drugs Today (Barc.) 2013, 49, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Haznedar, R. A new target for myeloma therapy. Blood 2012, 119, 1325–1326. [Google Scholar] [PubMed]
- Tripodo, C.; Florena, A.M.; Macor, P.; Di Bernardo, A.; Porcasi, R.; Guarnotta, C.; Ingrao, S.; Zerilli, M.; Secco, E.; Todaro, M.; et al. P-selectin glycoprotein ligand-1 as a potential target for humoral immunotherapy of multiple myeloma. Curr. Cancer Drug Targets 2009, 9, 617–625. [Google Scholar] [PubMed]
- Glavey, S.V.; Manier, S.; Natoni, A.; Sacco, A.; Moschetta, M.; Reagan, M.R.; Murillo, L.S.; Sahin, I.; Wu, P.; Mishima, Y.; et al. The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood 2014, 124, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Blau, O.; Baldus, C.D.; Hofmann, W.K.; Thiel, G.; Nolte, F.; Burmeister, T.; Turkmen, S.; Benlasfer, O.; Schumann, E.; Sindram, A.; et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011, 118, 5583–5592. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TKs | CAMs | ||
|---|---|---|---|
| Gene | Mutant Samples | Gene | Mutant Samples |
| c-KIT | 9.073 | PSGL1 | 140 |
| FLT3 | 17.467 | CXCR4 | 472 |
| JAK2 | 50.613 | CD34 | 119 |
| ABL1 | 1.961 | ITGα4 | 430 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Windisch, R.; Pirschtat, N.; Kellner, C.; Chen-Wichmann, L.; Lausen, J.; Humpe, A.; Krause, D.S.; Wichmann, C. Oncogenic Deregulation of Cell Adhesion Molecules in Leukemia. Cancers 2019, 11, 311. https://doi.org/10.3390/cancers11030311
Windisch R, Pirschtat N, Kellner C, Chen-Wichmann L, Lausen J, Humpe A, Krause DS, Wichmann C. Oncogenic Deregulation of Cell Adhesion Molecules in Leukemia. Cancers. 2019; 11(3):311. https://doi.org/10.3390/cancers11030311
Chicago/Turabian StyleWindisch, Roland, Nina Pirschtat, Christian Kellner, Linping Chen-Wichmann, Jörn Lausen, Andreas Humpe, Daniela S. Krause, and Christian Wichmann. 2019. "Oncogenic Deregulation of Cell Adhesion Molecules in Leukemia" Cancers 11, no. 3: 311. https://doi.org/10.3390/cancers11030311
APA StyleWindisch, R., Pirschtat, N., Kellner, C., Chen-Wichmann, L., Lausen, J., Humpe, A., Krause, D. S., & Wichmann, C. (2019). Oncogenic Deregulation of Cell Adhesion Molecules in Leukemia. Cancers, 11(3), 311. https://doi.org/10.3390/cancers11030311