Developing a Prognostic Gene Panel of Epithelial Ovarian Cancer Patients by a Machine Learning Model

 , ,
, ,
Abstract
1. Introduction
2. Results
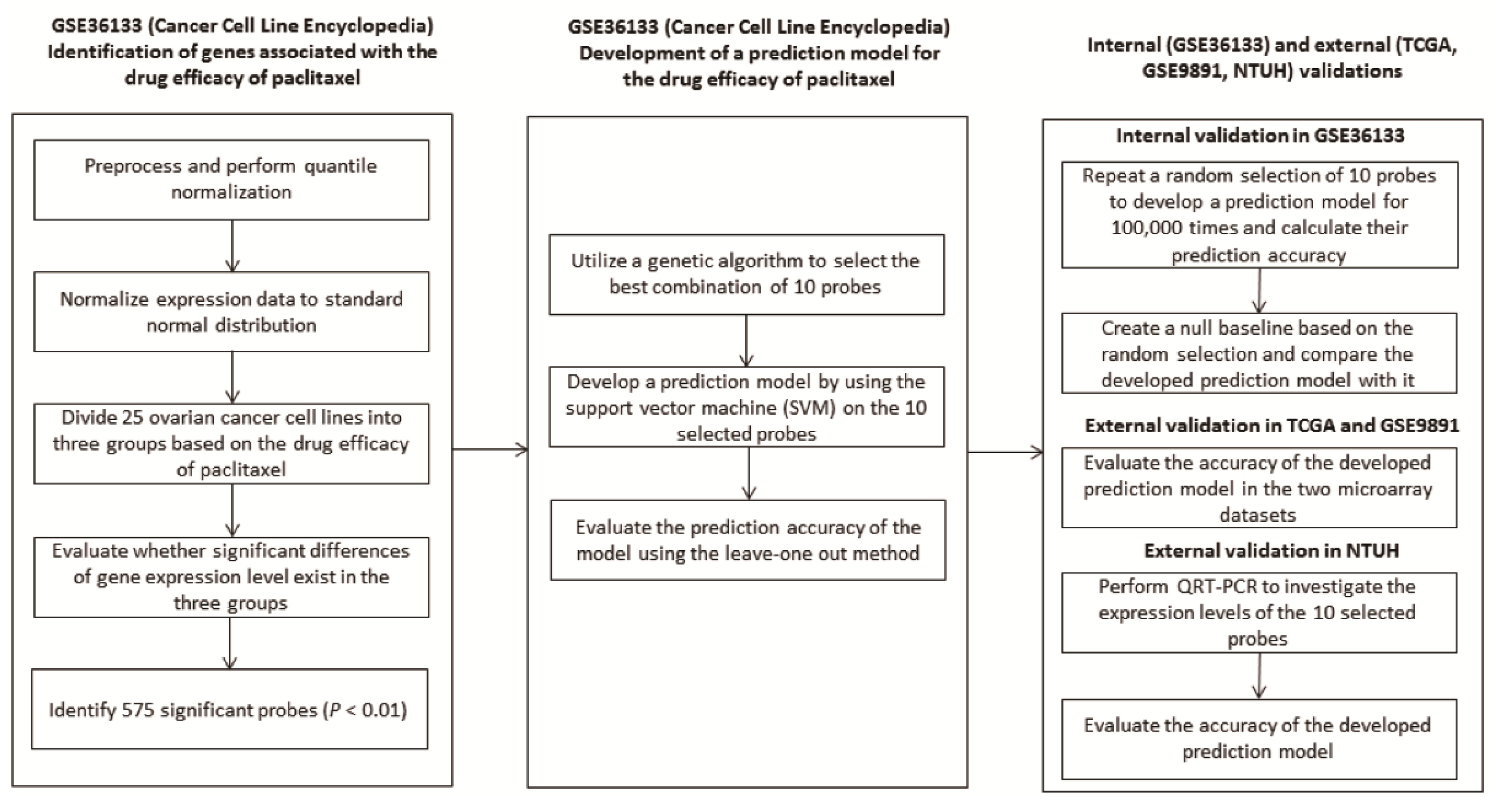
2.1. Development of A Predictive Model from Cancer Cell Line Encyclopaedia (CCLE)
2.2. Evaluation of The Performance of The Developed Predictive Model
2.3. Validation of The Predictive Model in Two Independent Datasets: TCGA and GSE9891
2.4. Validation of The Predictive Model by QRT-PCR in Ovarian Cancer Tissue of NTUH
3. Discussion
4. Materials and Methods
4.1. Identification of The Genes Associated with The Drug Response
4.2. Development of a Predictive Model Using a Genetic Algorithm
4.3. Validation of the Predictive Model in Two Independent Datasets
4.4. Validation of the Predictive Model by a Quantitative Real-Time Polymerase Chain Reaction (QRT-PCR) Using Ovarian Cancer Tissue
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.-C.; Chen, C.-A.; Chiang, C.-J.; Hsu, T.-H.; Lin, M.-C.; You, S.-L.; Cheng, W.-F.; Lai, M.-S. Trends in incidence and survival outcome of epithelial ovarian cancer: 30-year national population-based registry in Taiwan. J. Gynecol. Oncol. 2013, 24, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Monk, B.J.; Sood, A.K.; Herzog, T.J. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat. Rev. Clin. Oncol. 2013, 10, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Bottoni, P.; Scatena, R. The Role of CA 125 as Tumor Marker: Biochemical and Clinical Aspects. Adv. Exp. Med. Biol. 2015, 867, 229–244. [Google Scholar] [PubMed]
- Liu, J.; Westin, S.N. Rational selection of biomarker driven therapies for gynecologic cancers: The more we know, the more we know we don’t know. Gynecol. Oncol. 2016, 141, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.A.; Lai, L.C.; Tsai, M.H.; Lu, T.P.; Chuang, E.Y. Development of a prediction model for radiosensitivity using the expression values of genes and long non-coding RNAs. Oncotarget 2016, 7, 26739–26750. [Google Scholar] [CrossRef] [PubMed]
- Chuang, M.K.; Chiu, Y.C.; Chou, W.C.; Hou, H.A.; Chuang, E.Y.; Tien, H.F. A 3-microRNA scoring system for prognostication in de novo acute myeloid leukemia patients. Leukemia 2015, 29, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Ledermann, J.A.; Benedetti Panici, P. An overview of early investigational therapies for chemoresistant ovarian cancer. Expert Opin. Investig. Drugs 2015, 24, 1163–1183. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mahdi, H.; Bryant, C.; Shah, J.P.; Garg, G.; Munkarah, A. Clinical trials and progress with paclitaxel in ovarian cancer. Int. J. Womens Health 2010, 2, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Grendys, E.C., Jr.; Fiorica, J.V.; Orr, J.W., Jr.; Holloway, R.; Wang, D.; Tian, C.; Chan, J.K.; Herzog, T.J. Overview of a chemoresponse assay in ovarian cancer. Clin. Transl. Oncol. 2014, 16, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Au, K.K.; Josahkian, J.A.; Francis, J.A.; Squire, J.A.; Koti, M. Current state of biomarkers in ovarian cancer prognosis. Future Oncol. 2015, 11, 3187–3195. [Google Scholar] [CrossRef] [PubMed]
- Dorayappan, K.D.; Wallbillich, J.J.; Cohn, D.E.; Selvendiran, K. The biological significance and clinical applications of exosomes in ovarian cancer. Gynecol. Oncol. 2016, 142, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B. Recently identified drug resistance biomarkers in ovarian cancer. Expert Rev. Mol. Diagn. 2016, 16, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Alkema, N.G.; Wisman, G.B.; van der Zee, A.G.; van Vugt, M.A.; de Jong, S. Studying platinum sensitivity and resistance in high-grade serous ovarian cancer: Different models for different questions. Drug Resist. Updat. 2016, 24, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Huang, H.; Xiao, Z.; Zhang, W.; Cao, Y.; Qu, L.; Shou, C. Integrated analysis of gene expression profiles associated with response of platinum/paclitaxel-based treatment in epithelial ovarian cancer. PLoS ONE 2012, 7, e52745. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Matsumura, N.; Brown, J.B.; Wang, Z.; Yamaguchi, K.; Abiko, K.; Yoshioka, Y.; Hamanishi, J.; Baba, T.; Koshiyama, M.; et al. Prediction of taxane and platinum sensitivity in ovarian cancer based on gene expression profiles. Gynecol. Oncol. 2016, 141, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.P.; Chuang, E.Y.; Lu, T.P. Identification of predictive biomarkers for ZD-6474 in lung cancer. Transl. Cancer Res. 2015, 4, 324–331. [Google Scholar]
- Kim, S.W.; Kim, S.; Nam, E.J.; Jeong, Y.W.; Lee, S.H.; Paek, J.H.; Kim, J.H.; Kim, J.W.; Kim, Y.T. Comparative proteomic analysis of advanced serous epithelial ovarian carcinoma: Possible predictors of chemoresistant disease. OMICS 2011, 15, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.; Forrest, A.R.; Ehrlund, A.; Mejhert, N.; Itoh, M.; Kawaji, H.; Lassmann, T.; Laurencikiene, J.; Rydén, M.; Arner, P.; et al. Ceruloplasmin is a novel adipokine which is overexpressed in adipose tissue of obese subjects and in obesity-associated cancer cells. PLoS ONE 2014, 9, e80274. [Google Scholar] [CrossRef] [PubMed]
- Dentice, M.; Ambrosio, R.; Salvatore, D. Role of type 3 deiodinase in cancer. Expert Opin. Ther. Targets 2009, 13, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Ciavardelli, D.; Bellomo, M.; Crescimanno, C.; Vella, V. Type 3 deiodinase: Role in cancer growth, stemness, and metabolism. Front. Endocrinol. 2014, 5, 215. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Goubar, A.; Olaussen, K.A.; Vitale, I.; Senovilla, L.; Michels, J.; Robin, A.; Dorvault, N.; Besse, B.; Validire, P.; et al. Prognostic value of LIPC in non-small cell lung carcinoma. Cell Cycle 2013, 12, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Qin, Y.; Zhang, N.; Wang, J.; Wang, H.; Zheng, X. DISIS: Prediction of drug response through an iterative sure independence screening. PLoS ONE 2015, 10, e0120408. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Prat, J. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynaecol. Obstet. 2014, 124, 1–5. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Feature | Number (Proportion) |
|---|---|
| Total patients | 84 |
| Age (years) | 54.94 ± 11.25 |
| Histological subtype | |
| Serous | 59 (0.7) |
| Endometrioid | 12 (0.14) |
| Clear cell | 13 (0.16) |
| Tumor grade | |
| 1 | 4 (0.05) |
| 2 | 10 (0.12) |
| 3 | 57 (0.68) |
| Not available | 13 (0.15) |
| FIGO_stage | |
| Early (I + II) | 11 (0.13) |
| Advanced (III + IV) | 73 (0.87) |
| Debulking surgery | |
| Residual tumor size < 1 cm | 42 (0.5) |
| Residual tumor size ≥ 1 cm | 42 (0.5) |
| Feature | Numbers | Recurrence | Death | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | ||||||
| HR (95% CI) | p Value | HR (95% CI) | p Value | HR (95% CI) | p Value | HR (95% CI) | p Value | ||
| Predictive model | |||||||||
| Low | 19 | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| Medium + High | 65 | 0.643 (0.415–0.998) | 0.049 | 0.644 (0.436–0.952) | 0.027 | 0.559 (0.351–0.890) | 0.014 | 0.511 (0.334–0.783) | 0.002 |
| Residual tumor size < 1 cm | |||||||||
| No | 42 | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| Yes | 42 | 0.273 (0.160–0.468) | <0.001 | 0.312 (0.170–0.573) | <0.001 | 0.198 (0.107–0.365) | <0.001 | 0.252 (0.128–0.496) | <0.001 |
| FIGO stage | |||||||||
| Early (I + II) | 11 | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| Advanced (III + IV) | 73 | 7.954 (1.934–32.71) | 0.004 | 2.149 (0.257–17.93) | 0.480 | 6.13 (1.489–25.24) | 0.012 | 1.732 (0.201–14.90) | 0.617 |
| Tumor grade | |||||||||
| 1 | 4 | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| 2 + 3 | 67 | 2.289 (1.156–4.530) | 0.018 | 2.125 (0.992–4.552) | 0.053 | 1.756 (0.953–3.234) | 0.071 | 1.533 (0.761–3.09) | 0.232 |
| Histological subtype | |||||||||
| Serous | 59 | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| Endometrioid | 12 | 0.454 (0.193–1.067) | 0.070 | 1.023 (0.422–2.475) | 0.960 | 0.562 (0.239–1.323) | 0.187 | 1.077 (0.443–2.618) | 0.870 |
| Clear cell | 13 | 0.531 (0.239–1.180) | 0.120 | NA | NA | 0.284 (0.102–0.793) | 0.016 | NA | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, T.-P.; Kuo, K.-T.; Chen, C.-H.; Chang, M.-C.; Lin, H.-P.; Hu, Y.-H.; Chiang, Y.-C.; Cheng, W.-F.; Chen, C.-A. Developing a Prognostic Gene Panel of Epithelial Ovarian Cancer Patients by a Machine Learning Model. Cancers 2019, 11, 270. https://doi.org/10.3390/cancers11020270
Lu T-P, Kuo K-T, Chen C-H, Chang M-C, Lin H-P, Hu Y-H, Chiang Y-C, Cheng W-F, Chen C-A. Developing a Prognostic Gene Panel of Epithelial Ovarian Cancer Patients by a Machine Learning Model. Cancers. 2019; 11(2):270. https://doi.org/10.3390/cancers11020270
Chicago/Turabian StyleLu, Tzu-Pin, Kuan-Ting Kuo, Ching-Hsuan Chen, Ming-Cheng Chang, Hsiu-Ping Lin, Yu-Hao Hu, Ying-Cheng Chiang, Wen-Fang Cheng, and Chi-An Chen. 2019. "Developing a Prognostic Gene Panel of Epithelial Ovarian Cancer Patients by a Machine Learning Model" Cancers 11, no. 2: 270. https://doi.org/10.3390/cancers11020270
APA StyleLu, T.-P., Kuo, K.-T., Chen, C.-H., Chang, M.-C., Lin, H.-P., Hu, Y.-H., Chiang, Y.-C., Cheng, W.-F., & Chen, C.-A. (2019). Developing a Prognostic Gene Panel of Epithelial Ovarian Cancer Patients by a Machine Learning Model. Cancers, 11(2), 270. https://doi.org/10.3390/cancers11020270