Decoration of Anti-CD38 on Nanoparticles Carrying a STAT3 Inhibitor Can Improve the Therapeutic Efficacy Against Myeloma

 ,
, 
Abstract
1. Introduction
2. Results
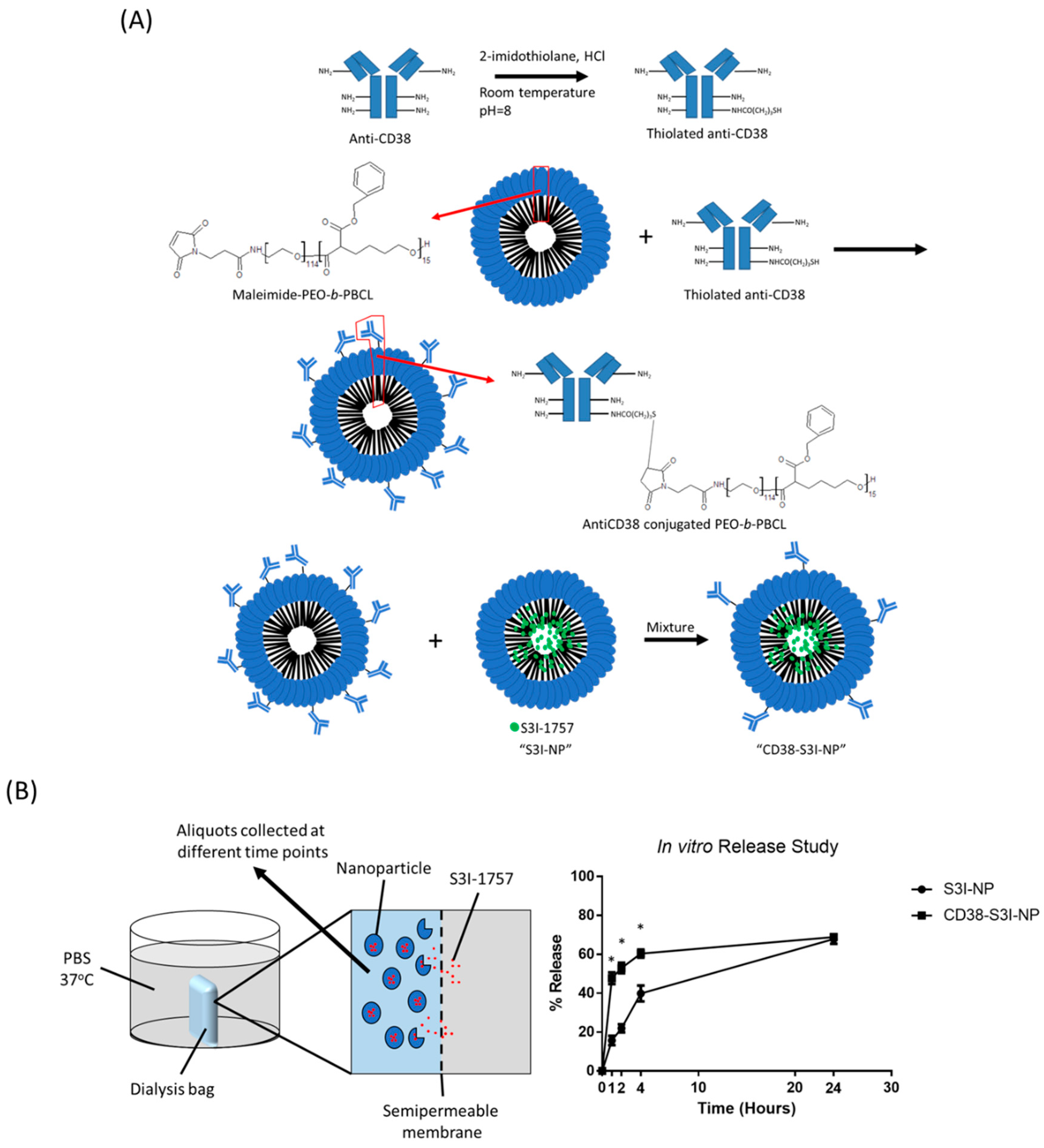
2.1. Synthesis and Characterization of CD38-S3I-NP
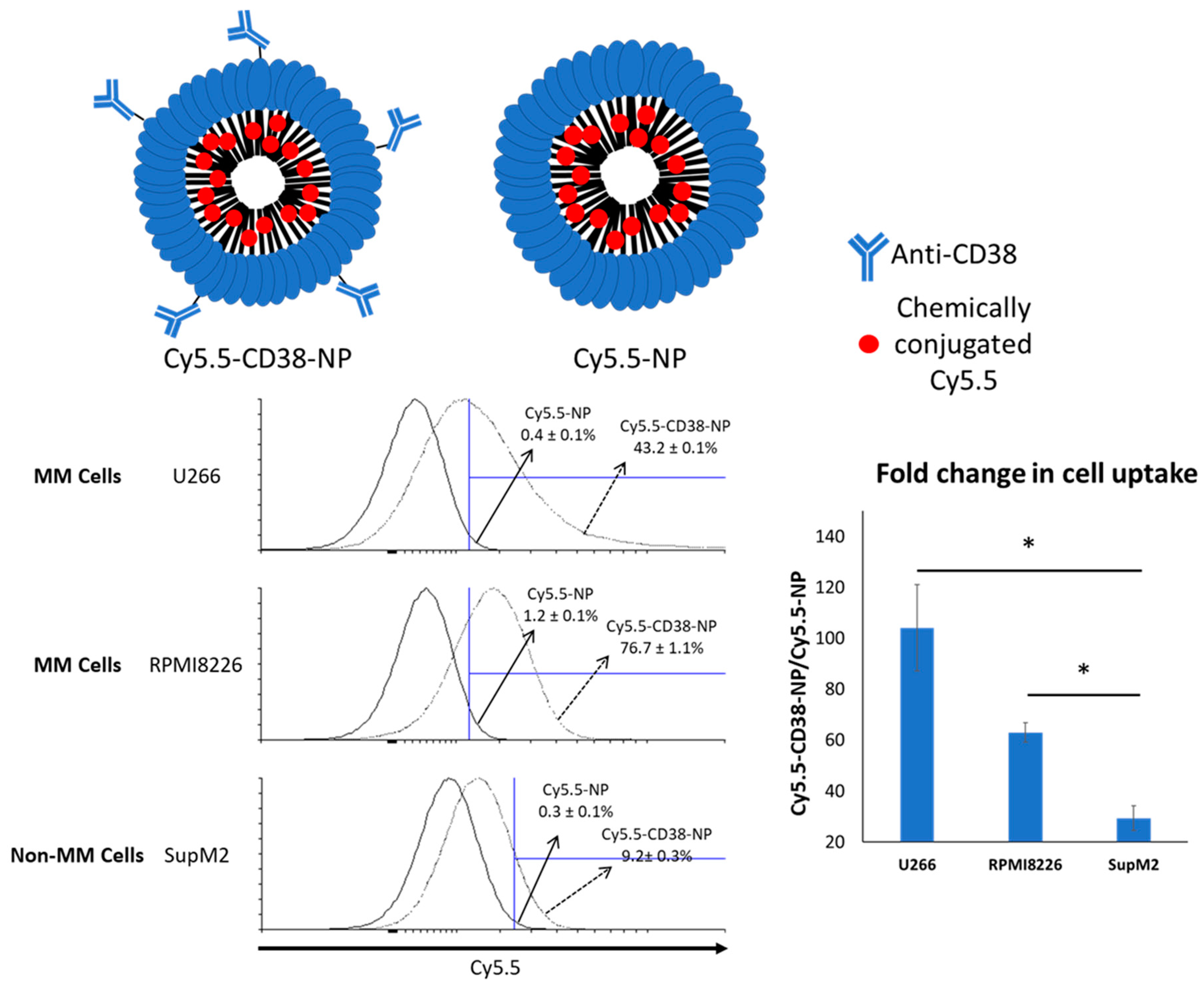
2.2. Anti-CD38 Conjugation on NP Results in More Cellular Uptake by MM Cells
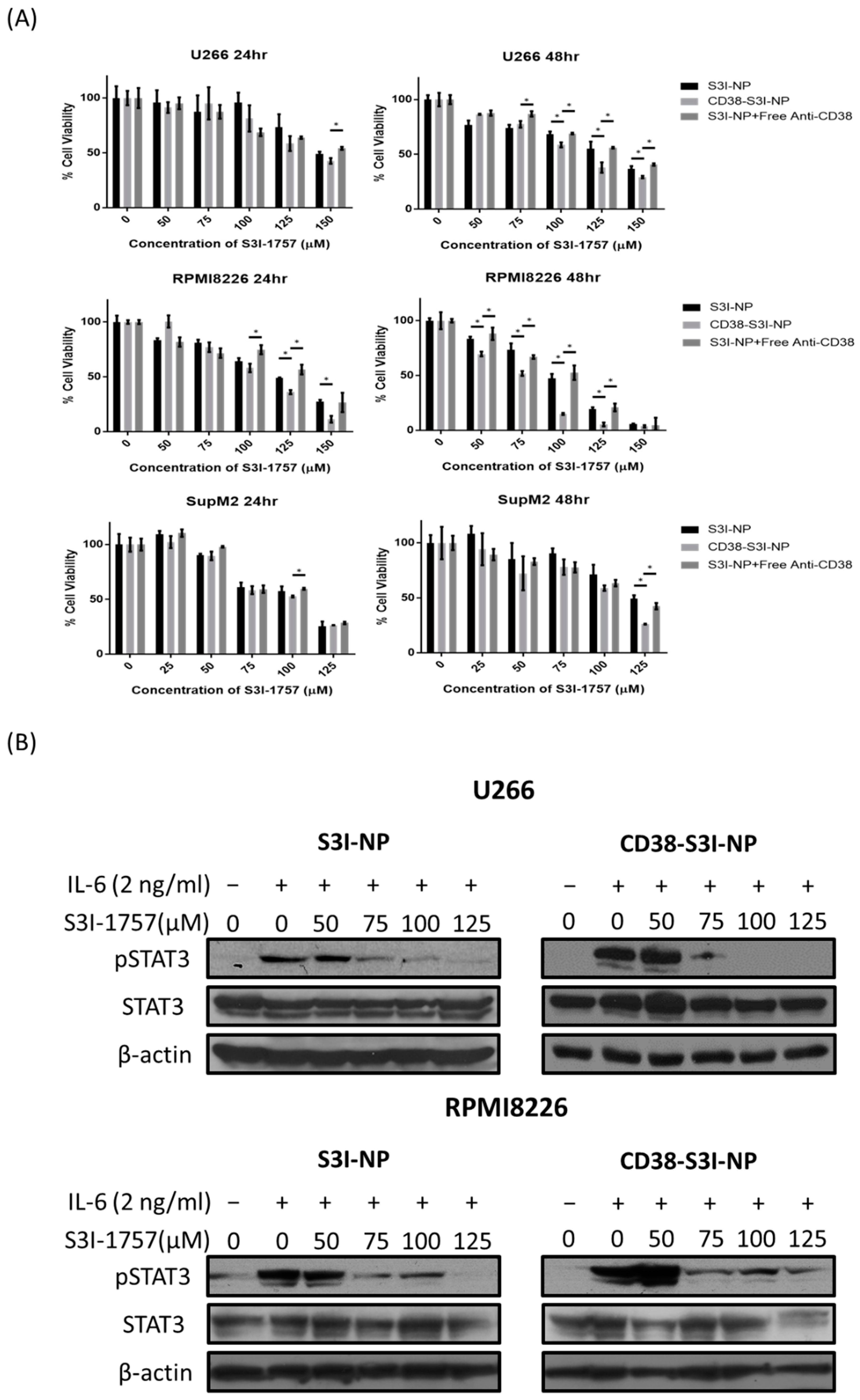
2.3. CD38-S3I-NP is More Cytotoxic to MM Cells than S3I-NP
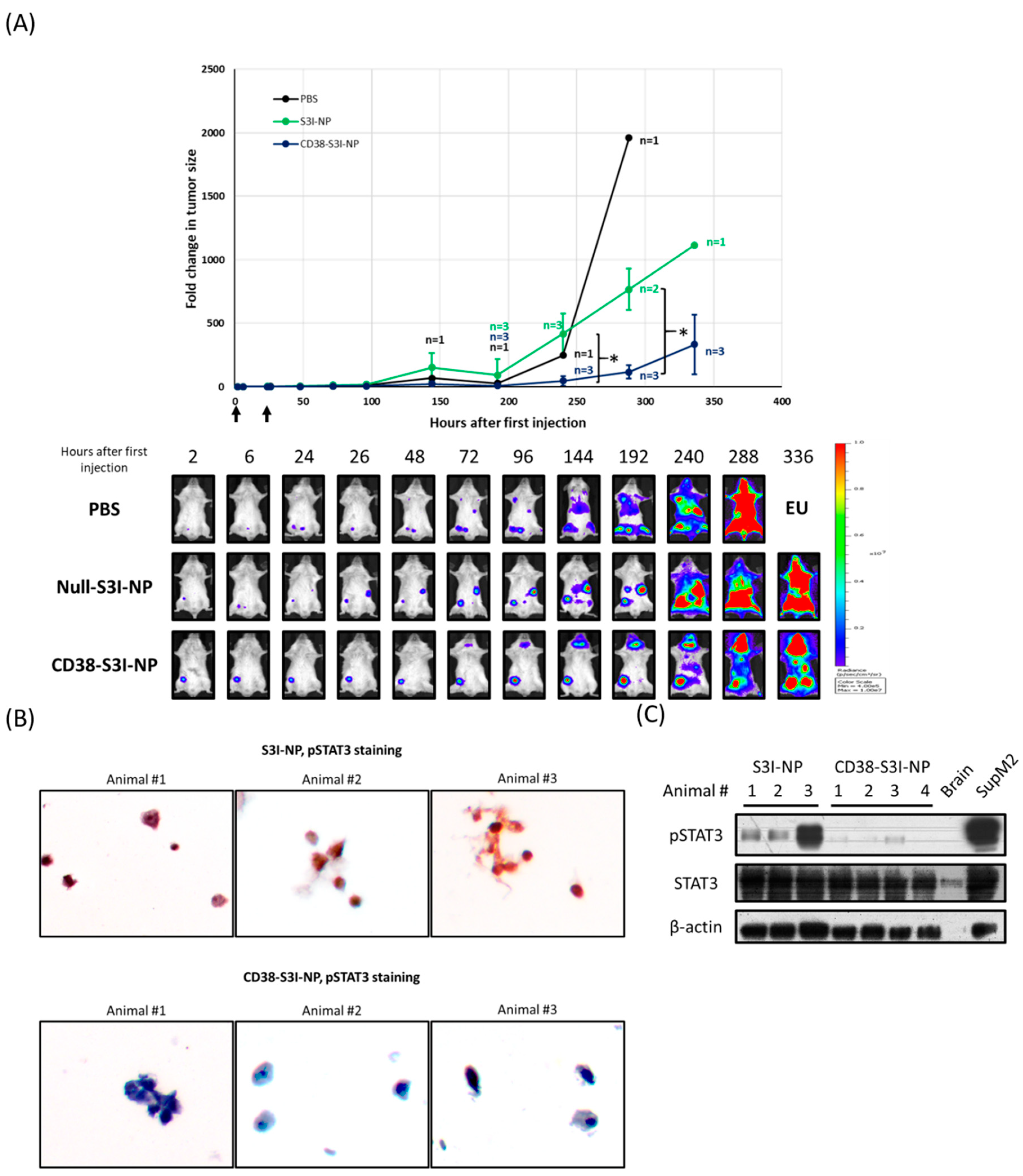
2.4. CD38-S3I-NP is More Effective in Suppressing MM Tumor Growth In Vivo Compared to S3I-NP
3. Discussion
4. Materials and Methods
4.1. Materials and Cell Culture
4.2. Purification of Anti-CD38
4.3. Preparation of NP
4.4. In Vitro Release Assay
4.5. Cellular Uptake Assay
4.6. Cell Viability Assay
4.7. Western Blot Analysis
4.8. Preparation of U266-luc Cells by Lentiviral Transduction
4.9. In Vivo Studies Using MM Xenograft
4.10. Immunocytochemistry
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Arruebo, M.; Valladares, M.; González-Fernández, Á. Antibody-conjugated nanoparticles for biomedical applications. J. Nanomater. 2009, 2009, 439389. [Google Scholar] [CrossRef]
- Mi, Y.; Liu, X.; Zhao, J.; Ding, J.; Feng, S.S. Multimodality treatment of cancer with herceptin conjugated, thermomagnetic iron oxides and docetaxel loaded nanoparticles of biodegradable polymers. Biomaterials 2012, 33, 7519–7529. [Google Scholar] [CrossRef]
- Parhi, P.; Sahoo, S.K. Trastuzumab guided nanotheranostics: A lipid based multifunctional nanoformulation for targeted drug delivery and imaging in breast cancer therapy. J. Colloid Interface Sci. 2015, 451, 198–211. [Google Scholar] [CrossRef]
- Vivek, R.; Thangam, R.; NipunBabu, V.; Rejeeth, C.; Sivasubramanian, S.; Gunasekaran, P.; Muthuchelian, K.; Kannan, S. Multifunctional HER2-Antibody Conjugated Polymeric Nanocarrier-Based Drug Delivery System for Multi-Drug-Resistant Breast Cancer Therapy. ACS Appl. Mater. Interfaces 2014, 6, 6469–6480. [Google Scholar] [CrossRef]
- Choi, W.I.; Lee, J.H.; Kim, J.Y.; Heo, S.U.; Jeong, Y.Y.; Kim, Y.H.; Tae, G. Targeted antitumor efficacy and imaging via multifunctional nano-carrier conjugated with anti-HER2 trastuzumab. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 359–368. [Google Scholar] [CrossRef]
- Punfa, W.; Yodkeeree, S.; Pitchakarn, P.; Ampasavate, C.; Limtrakul, P. Enhancement of cellular uptake and cytotoxicity of curcumin-loaded PLGA nanoparticles by conjugation with anti-P-glycoprotein in drug resistance cancer cells. Acta Pharmacol. Sin. 2012, 33, 823–831. [Google Scholar] [CrossRef]
- Arya, G.; Vandana, M.; Acharya, S.; Sahoo, S.K. Enhanced antiproliferative activity of Herceptin (HER2)-conjugated gemcitabine-loaded chitosan nanoparticle in pancreatic cancer therapy. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 859–870. [Google Scholar] [CrossRef]
- Sreeranganathan, M.; Uthaman, S.; Sarmento, B.; Mohan, C.G.; Park, I.K.; Jayakumar, R. In vivo evaluation of cetuximab-conjugated poly(γ-glutamic acid)-docetaxel nanomedicines in EGFR-overexpressing gastric cancer xenografts. Int. J. Nanomed. 2017, 12, 7167–7182. [Google Scholar] [CrossRef]
- Mukerjee, A.; Ranjan, A.P.; Vishwanatha, J.K. Targeted nanocurcumin therapy using annexin A2 antibody improves tumor accumulation and therapeutic efficacy against highly metastatic breast cancer. J. Biomed. Nanotechnol. 2016, 12, 1374–1392. [Google Scholar] [CrossRef]
- Öztürk, K.; Esendağlı, G.; Gürbüz, M.U.; Tülü, M.; Çalış, S. Effective targeting of gemcitabine to pancreatic cancer through PEG-cored Flt-1 antibody-conjugated dendrimers. Int. J. Pharm. 2017, 517, 157–167. [Google Scholar] [CrossRef]
- Punfa, W.; Suzuki, S.; Pitchakarn, P.; Yodkeeree, S.; Naiki, T.; Takahashi, S.; Limtrakul, P. Curcumin-loaded PLGA nanoparticles conjugated with anti-P-glycoprotein antibody to overcome multidrug resistance. Asian Pac. J. Cancer Prev. 2014, 15, 9249–9258. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Akita, H.; Ishida, E.; Hashimoto, K.; Kobayashi, H.; Aoki, T.; Yasuda, J.; Obata, K.; Kikuchi, H.; Ishida, T.; et al. Tumor targeting of doxorubicin by anti-MT1-MMP antibody-modified PEG liposomes. Int. J. Pharm. 2007, 342, 194–200. [Google Scholar] [CrossRef]
- Sun, T.; Wu, H.; Li, Y.; Huang, Y.; Yao, L.; Chen, X.; Han, X.; Zhou, Y.; Du, Z. Targeting transferrin receptor delivery of temozolomide for a potential glioma stem cell-mediated therapy. Oncotarget 2017, 8, 74451–74465. [Google Scholar] [CrossRef]
- Bharti, A.C.; Shishodia, S.; Reuben, J.M.; Weber, D.; Alexanian, R.; Raj-Vadhan, S.; Estrov, Z.; Talpaz, M.; Aggarwal, B.B. Nuclear factor–κB and STAT3 are constitutively active in CD138 + cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood 2004, 103, 3175–3184. [Google Scholar] [CrossRef]
- Liu, T.; Fei, Z.; Gangavarapu, K.J.; Agbenowu, S.; Bhushan, A.; Lai, J.C.; Daniels, C.K.; Cao, S. Interleukin-6 and JAK2/STAT3 signaling mediate the reversion of dexamethasone resistance after dexamethasone withdrawal in 7TD1 multiple myeloma cells. Leuk. Res. 2013, 37, 1322–1328. [Google Scholar] [CrossRef]
- Zhang, X.D.; Baladandayuthapani, V.; Lin, H.; Mulligan, G.; Li, B.-Z.; Esseltine, D.L.W.; Qi, L.; Xu, J.; Hunziker, W.; Barlogie, B.; et al. Tight jnction protein 1 modulates proteasome capacity and proteasome inhibitor sensitivity in multiple myeloma via EGFR/JAK1/STAT3 signaling. Cancer Cell 2016, 29, 639–652. [Google Scholar] [CrossRef]
- Wu, W.; Ma, D.; Wang, P.; Cao, L.; Lu, T.; Fang, Q.; Zhao, J.; Wang, J. Potential crosstalk of the interleukin-6-heme oxygenase-1-dependent mechanism involved in resistance to lenalidomide in multiple myeloma cells. FEBS J. 2016, 283, 834–849. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Pireddu, R.; Yang, H.; Urlam, M.K.; Lawrence, H.R.; Guida, W.C.; Lawrence, N.J.; Sebti, S.M. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013, 73, 1922–1933. [Google Scholar] [CrossRef]
- Zhang, X.; Yue, P.; Fletcher, S.; Zhao, W.; Gunning, P.T.; Turkson, J. A novel small-molecule disrupts Stat3 SH2 domain–phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem. Pharmacol. 2010, 79, 1398–1409. [Google Scholar] [CrossRef]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Lee, S.-H.; Han, S.-W.; Kim, M.-J.; Kim, T.-M.; Kim, T.-Y.; Heo, D.S.; Yuasa, M.; Yanagihara, Y.; Bang, Y.J. Phase I Study of OPB-31121, an Oral STAT3 Inhibitor, in Patients with Advanced Solid Tumors. Cancer Res. Treat. 2015, 47, 607–615. [Google Scholar] [CrossRef]
- Soleimani, A.H.; Garg, S.M.; Paiva, I.M.; Vakili, M.R.; Alshareef, A.; Huang, Y.-H.; Molavi, O.; Lai, R.; Lavasanifar, A. Micellar nano-carriers for the delivery of STAT3 dimerization inhibitors to melanoma. Drug Deliv. Transl. Res. 2017, 1–11. [Google Scholar] [CrossRef]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug Delivery: Is the Enhanced Permeability and Retention Effect Sufficient for Curing Cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef]
- Martin, C.; Aibani, N.; Callan, J.F.; Callan, B. Recent advances in amphiphilic polymers for simultaneous delivery of hydrophobic and hydrophilic drugs. Ther. Deliv. 2016, 7, 15–31. [Google Scholar] [CrossRef]
- Molavi, O.; Ma, Z.; Mahmud, A.; Alshamsan, A.; Samuel, J.; Lai, R.; Kwon, G.S.; Lavasanifar, A. Polymeric micelles for the solubilization and delivery of STAT3 inhibitor cucurbitacins in solid tumors. Int. J. Pharm. 2008, 347, 118–127. [Google Scholar] [CrossRef]
- Wang, X.B.; Zhou, H.Y. Molecularly targeted gemcitabine-loaded nanoparticulate system towards the treatment of EGFR overexpressing lung cancer. Biomed. Pharmacother. 2015, 70, 123–128. [Google Scholar] [CrossRef]
- Karra, N.; Nassar, T.; Ripin, A.N.; Schwob, O.; Borlak, J.; Benita, S. Antibody conjugated PLGA nanoparticles for targeted delivery of paclitaxel palmitate: Efficacy and biofate in a lung cancer mouse model. Small 2013, 9, 4221–4236. [Google Scholar] [CrossRef]
- Molavi, O.; Xiong, X.B.; Douglas, D.; Kneteman, N.; Nagata, S.; Pastan, I.; Chu, Q.; Lavasanifar, A.; Lai, R. Anti-CD30 antibody conjugated liposomal doxorubicin with significantly improved therapeutic efficacy against anaplastic large cell lymphoma. Biomaterials 2013, 34, 8718–8725. [Google Scholar] [CrossRef]
- Badkas, A.; Frank, E.; Zhou, Z.; Jafari, M.; Chandra, H.; Sriram, V.; Lee, J.Y.; Yadav, J.S. Modulation of in vitro phagocytic uptake and immunogenicity potential of modified Herceptin®-conjugated PLGA-PEG nanoparticles for drug delivery. Colloids Surf. B Biointerfaces 2018, 162, 271–278. [Google Scholar] [CrossRef]
- Lin, L.; Benson, D.M.; Deangelis, S.; Bakan, C.E.; Li, P.K.; Li, C.; Lin, J. A small molecule, LLL12 inhibits constitutive STAT3 and IL-6-induced STAT3 signaling and exhibits potent growth suppressive activity in human multiple myeloma cells. Int. J. Cancer 2012, 130, 1459–1469. [Google Scholar] [CrossRef]
- Zhang, Z.; Mao, H.; Du, X.; Zhu, J.; Xu, Y.; Wang, S. A novel small molecule agent displays potent anti-myeloma activity by inhibiting the JAK2-STAT3 signaling pathway. Oncotarget 2016, 7, 9296–9308. [Google Scholar] [CrossRef]
- Xiang, M.; Kim, H.; Ho, V.T.; Walker, S.R.; Bar-Natan, M.; Anahtar, M.; Liu, S.; Toniolo, P.A.; Kroll, Y.; Jones, N.; et al. Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood 2016, 128, 1845–1853. [Google Scholar] [CrossRef]
- Malavasi, F.; Funaro, A.; Alessio, M.; DeMonte, L.B.; Ausiello, C.M.; Dianzani, U.; Lanza, F.; Magrini, E.; Momo, M.; Roggero, S. CD38: A multi-lineage cell activation molecule with a split personality. Int. J. Clin. Lab. Res. 1992, 22, 73–80. [Google Scholar] [CrossRef]
- De Weers, M.; Tai, Y.-T.; van derVeer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Beum, P.V.; Lindorfer, M.A.; Peek, E.M.; Stukenberg, P.T.; deWeers, M.; Beurskens, F.J.; Parren, P.W.; van de Winkel, J.G.; Taylor, R.P. Penetration of antibody-opsonized cells by the membrane attack complex of complement promotes Ca2+ influx and induces streamers. Eur. J. Immunol. 2011, 41, 2436–2446. [Google Scholar] [CrossRef]
- Dela Puente, P.; Luderer, M.J.; Federico, C.; Jin, A.; Gilson, R.C.; Egbulefu, C.; Alhallak, K.; Shah, S.; Muz, B.; Sun, J.; et al. Enhancing proteasome-inhibitory activity and specificity of bortezomib by CD38 targeted nanoparticles in multiple myeloma. J. Control. Release 2018, 270, 158–176. [Google Scholar] [CrossRef]
- Mahmud, A.; Xiong, X.B.; Lavasanifar, A. Novel self-associating poly(ethylene oxide)-block-poly(ε-caprolactone) block copolymers with functional side groups on the polyester block for drug delivery. Macromolecules 2006, 39, 9419–9428. [Google Scholar] [CrossRef]
- Kogan, T. The synthesis of substituted methoxy-poly (ethyleneglycol) derivatives suitable for selective protein modification. Synth. Commun. 1992, 22, 2417–2424. [Google Scholar] [CrossRef]
- Garg, S.M.; Paiva, I.M.; Vakili, M.R.; Soudy, R.; Agopsowicz, K.; Soleimani, A.H.; Hitt, M.; Kaur, K.; Lavasanifar, A. Traceable PEO-poly(ester) micelles for breast cancer targeting: The effect of core structure and targeting peptide on micellar tumor accumulation. Biomaterials 2017, 144, 17–29. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NP Formulation | Average Size (nm) | Polydispersity Index | Drug Encapsulation Efficiency (%) | Drug Loading (Weight %) |
|---|---|---|---|---|
| S3I-NP | 97.4 ± 5.2 | 0.273 ± 0.003 | 87.0 ± 9.2% | 15.7 ± 1.7% |
| CD38-S3I-NP | 91.4 ± 9.4 | 0.367 ± 0.016* | 81.6 ± 7.2% | 14.7 ± 1.3% |
| Cell Line | Treatment | IC50 (μM) | |
|---|---|---|---|
| 24 h | 48 h | ||
| U266 | S3I-NP | 136.7–163.6 | 115.4–148.6 ** |
| CD38-S3I-NP | 127.7–151.3 | 106.3–114.0 | |
| S3I-NP + Free Anti-CD38 | 143.5–172.4 | 128.6–139.4 ** | |
| RPMI8226 | S3I-NP | 110.9–124.5 | 88.2–98.1 ** |
| CD38-S3I-NP | 100.4–109.8 | 64.0–73.6 | |
| S3I-NP + Free Anti-CD38 | 108.9–142.2 | 87.2–99.5 ** | |
| SupM2 | S3I-NP | 88.8–105.6 | 110.4–144.8 |
| CD38-S3I-NP | 86.0–98.9 | 83.16–119.4 | |
| S3I-NP + Free Anti-CD38 | 90.1–110.0 | 106.9–134.9 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-H.; Vakili, M.R.; Molavi, O.; Morrissey, Y.; Wu, C.; Paiva, I.; Soleimani, A.H.; Sanaee, F.; Lavasanifar, A.; Lai, R. Decoration of Anti-CD38 on Nanoparticles Carrying a STAT3 Inhibitor Can Improve the Therapeutic Efficacy Against Myeloma. Cancers 2019, 11, 248. https://doi.org/10.3390/cancers11020248
Huang Y-H, Vakili MR, Molavi O, Morrissey Y, Wu C, Paiva I, Soleimani AH, Sanaee F, Lavasanifar A, Lai R. Decoration of Anti-CD38 on Nanoparticles Carrying a STAT3 Inhibitor Can Improve the Therapeutic Efficacy Against Myeloma. Cancers. 2019; 11(2):248. https://doi.org/10.3390/cancers11020248
Chicago/Turabian StyleHuang, Yung-Hsing, Mohammad Reza Vakili, Ommoleila Molavi, Yuen Morrissey, Chengsheng Wu, Igor Paiva, Amir Hasan Soleimani, Forugh Sanaee, Afsaneh Lavasanifar, and Raymond Lai. 2019. "Decoration of Anti-CD38 on Nanoparticles Carrying a STAT3 Inhibitor Can Improve the Therapeutic Efficacy Against Myeloma" Cancers 11, no. 2: 248. https://doi.org/10.3390/cancers11020248
APA StyleHuang, Y.-H., Vakili, M. R., Molavi, O., Morrissey, Y., Wu, C., Paiva, I., Soleimani, A. H., Sanaee, F., Lavasanifar, A., & Lai, R. (2019). Decoration of Anti-CD38 on Nanoparticles Carrying a STAT3 Inhibitor Can Improve the Therapeutic Efficacy Against Myeloma. Cancers, 11(2), 248. https://doi.org/10.3390/cancers11020248