Standardization of Somatic Variant Classifications in Solid and Haematological Tumours by a Two-Level Approach of Biological and Clinical Classes: An Initiative of the Belgian ComPerMed Expert Panel

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Expert Panel
2.2. Variant Calling and Annotation
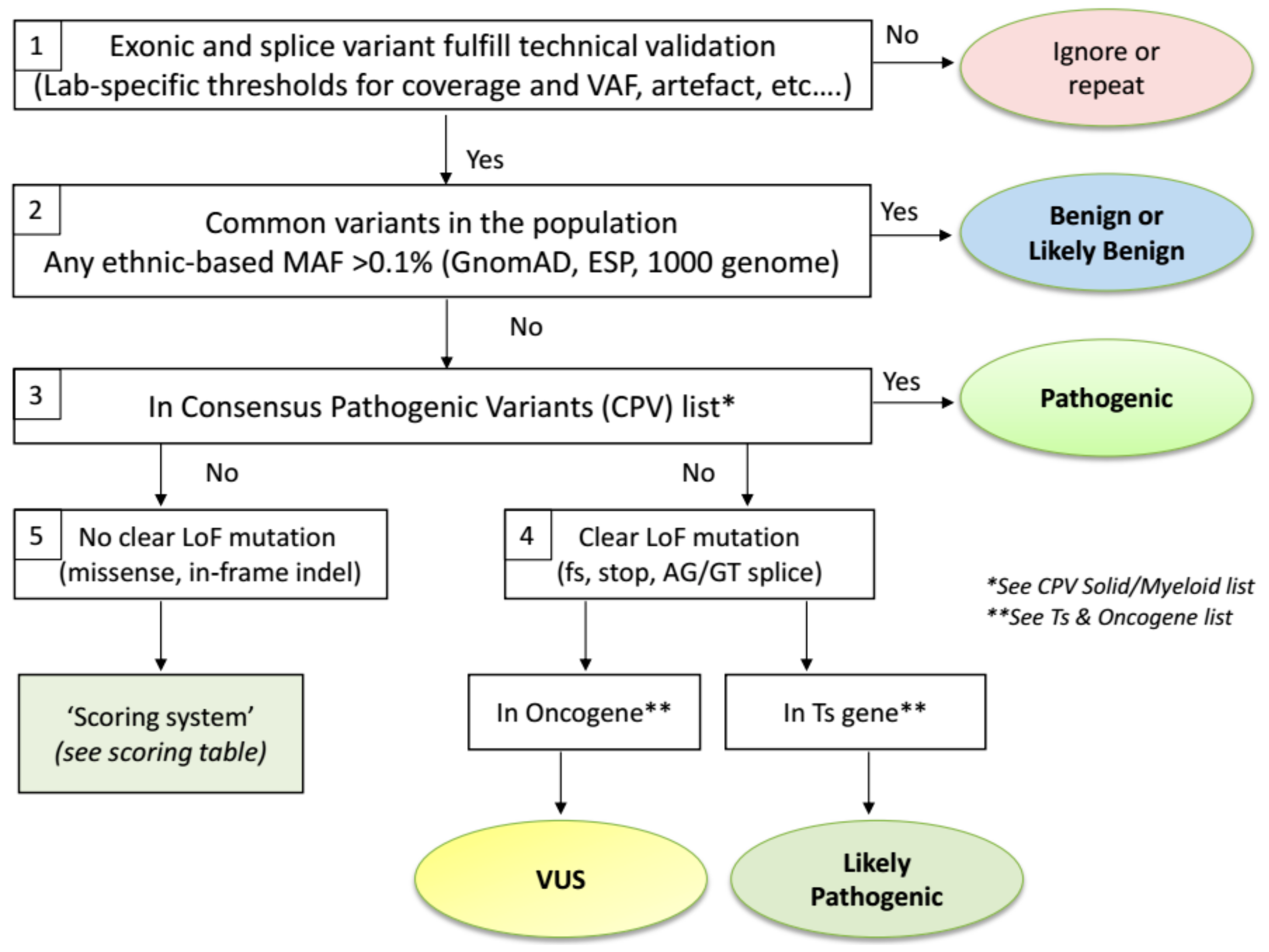
2.3. Biological Variant Classification Workflow
2.4. Exceptions to the Workflow
- The Ts gene TP53 is an exceptional gene because of the plethora of variants detected in many tumour types affecting almost every position of the p53 protein. Therefore, we advise to use two dedicated TP53 databases to assess variant pathogenicity. The International Agency for Research in Cancer (IARC) TP53 database (http://p53.iarc.fr/) compiles various types of information on human TP53 variations in relation to cancer [26]. The second database, Seshat (http://vps338341.ovh.net/), can be used for (predicted) functional consequences of protein changes. The tumour-related outcome is presented in the downloadable Summary report. The consensus class indicated by both tools will be used for TP53 variant classification. However, in comparison with the ERIC recommendations [27] that classify the −2, −1 and +1, +2 exon flanking splice variants as Pathogenic, we mark them as Likely Pathogenic since these variants are actually not different from frameshift variants that are also classified as Likely Pathogenic by ERIC. Secondly, synonymous changes that are predicted to affect splicing are classified as Pathogenic by ERIC. Importantly, synonymous changes in P53 should also be checked for a detrimental functional effect in both TP53 databases.
- The BRCA1 and BRCA2 Ts genes are specifically analysed for variants in gynaecological tumours of the ovary and endometrium, as well as in cancers from breast, pancreas and prostate. In these cases, clear LoF variants (nonsense, frameshift, splice sites) are always classified as Pathogenic, instead of Likely Pathogenic if classified via the ComPerMed workflow. Notably, LoF variants in the last exon as well as all other somatic variants need to be checked for their pathogenicity in different online databases including ARUP (http://www.arup.utah.edu/database/BRCA/), InterVar (http://wintervar.wglab.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), Enigma (https://brcaexchange.org/) and LOVD (https://databases.lovd.nl/shared/genes).
- Sequencing stutters of short tandem repeats (STR), including homopolymers, often occur as sequencing errors that are present in Vcf files at low allele frequencies, typically lower than 5%. However, a true change at an STR site can be discriminated from a stutter if the VAF is significantly higher than the observed stutter error rate present in most samples. The VAF of each STR variant thus has to be checked and if higher than a validated lab-specific threshold, it should be regarded as a true event. This threshold has to be defined by each lab since it can be method or analysis-specific. Each true STR change has to follow the standard classification workflow. As the prime example, the frameshift c.1934dup in the Ts gene ASXL1 is often seen as a stutter error in many NGS workflows at VAFs up to 10% but can be also found as a true variant in AML samples with VAF’s above the lab-specific threshold, thus classifying it as Likely Pathogenic.
- Splice site variants should be restricted to the −2, −1 and +1, +2 exon flanking positions, which harbour the AG/GT consensus splice motif, except for the MET exon 14 and BRCA1 and BRCA2 splice regions that should be analysed more broadly. All splice site variants will be considered as loss-of-function variants (Likely Pathogenic class) even though it might result in an in-frame exon(s) deletion because loss of at least one exon will most likely functionally harm the protein as well.
- Splice site variants in exon 14 of MET have to be seen as a CPV and thus are biologically classified as Pathogenic.
- Out-of-frame indels in exon 9 of CALR, including the typical type I and type II mutations, as well as out-of-frame insertions in exon 11 of NPM1 should not be treated as frameshift variants but as Consensus Pathogenic Variants (CPVs). Therefore, they are classified as Pathogenic.
- Somatic in-frame indels in the bZIP domain of CEBPA should be regarded as Likely Pathogenic.
- Finally, population-specific very rare benign variants can be distinguished from true somatic variants by their presence in at least three region-specific samples, with VAFs close to 50%, irrespective of the tumour content or tumour type. Consultancy of neighbouring NGS labs is advised and follow-up of such Likely Benign variants is warranted.
2.5. The Clinical Report
3. Discussion
- (1)
- The minor allele frequency (MAF) threshold to assign a variant as a polymorphism (class Likely Benign or Benign) was set at 1% by ACMG and GESMD, which is especially important in the absence of paired normal tissue. We have lowered this threshold to 0.1% because of the much higher number of data since 2015, and the curation of population databases thereby minimising the contamination of somatic tumour variants. Moreover, this threshold is valid for ethnic-specific MAFs with at least 2000 alleles investigated [42], which can be consulted in gnomAD. Note however, that this MAF threshold can be influenced by the targeted NGS method employed [43]. Finally, variants in ASXL1, DNMT3A and TET2 with VAFs below 10% can be associated with Clonal Hematopoiesis of Indeterminate Potential (CHIP) [44] and thus should be interpreted with caution as their presence alone is no evidence for the presence of malignancy;
- (2)
- ACMG advises to use the genomic coordinates of variants to be able to query genomic databases and not to depend on transcripts that are prone to changes. We recommend to use the HGVS nomenclature with reference to the transcript ID with the NCBI accession number of the main transcript, with version (e.g., BRAF NM_004333.5). We anticipate to change to the Locus Reference Genomic (LRG) record (http://www.lrg-sequence.org/) as it contains a stable reference sequence. So far, not all genes acquired an LRG number, and since most annotation programs and databases do not yet include the LRG transcript numbers, we did not make this switch yet;
- (3)
- For splice variants, we only consider the intronic −2, −1 (AG) and +1, +2 (GT) consensus splice positions, except MET exon 14 and BRACA1/2. ACMG and GESMD also evaluates intronic and exonic variants in the proximity of the splice sites, which are subjected to in-silico splice prediction tools. However, because of the low specificity of these tools and the inherent requirement for functional confirmation, we are not in favour of this option;
- (4)
- We classify the LoF variants in the last exon in the same way as those in preceding exons. GESPD requires the further evaluation of these changes.
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Luthra, R.; Chen, H.; Roy-Chowdhuri, S.; Singh, R.R. Next-Generation Sequencing in Clinical Molecular Diagnostics of Cancer: Advantages and Challenges. Cancers 2015, 7, 2023–2036. [Google Scholar] [CrossRef]
- Baeissa, H.M.; Benstead-Hume, G.; Richardson, C.J.; Pearl, F.M. Mutational patterns in oncogenes and tumour suppressors. Biochem. Soc. Trans. 2016, 44, 925–931. [Google Scholar] [CrossRef]
- Llinas-Arias, P.; Esteller, M. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: An update. Open Biol. 2017, 7, 170152. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Frohling, S.; Glimm, H. Integrating next-generation sequencing into clinical oncology: Strategies, promises and pitfalls. ESMO Open 2016, 1, e000094. [Google Scholar] [CrossRef] [PubMed]
- Vanderwalde, A.; Spetzler, D.; Xiao, N.; Gatalica, Z.; Marshall, J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018, 7, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Kameyama, H.; Takabe, K.; Okuda, S.; Wakai, T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019, 110, 6–15. [Google Scholar] [CrossRef]
- Kumar, S.; Razzaq, S.K.; Vo, A.D.; Gautam, M.; Li, H. Identifying fusion transcripts using next generation sequencing. Wiley Interdiscip. Rev. RNA 2016, 7, 811–823. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Stojanov, P.; Perrin, D.L.; Cibulskis, K.; Marlow, S.; Jane-Valbuena, J.; Friedrich, D.C.; Kryukov, G.; Carter, S.L.; et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 2014, 20, 682–688. [Google Scholar] [CrossRef]
- Sukhai, M.A.; Craddock, K.J.; Thomas, M.; Hansen, A.R.; Zhang, T.; Siu, L.; Bedard, P.; Stockley, T.L.; Kamel-Reid, S. A classification system for clinical relevance of somatic variants identified in molecular profiling of cancer. Genet. Med. 2016, 18, 128–136. [Google Scholar] [CrossRef]
- Hoskinson, D.C.; Dubuc, A.M.; Mason-Suares, H. The current state of clinical interpretation of sequence variants. Curr. Opin. Genet. Dev. 2017, 42, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.K.Y.; Bedard, P.L.; Tortora, G.; Douillard, J.Y.; et al. A framework to rank genomic alterations as targets for cancer precision medicine: The ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Klink, B.; Heining, C.; Groschel, S.; Hutter, B.; Frohlich, M.; Uhrig, S.; Hubschmann, D.; Schlesner, M.; Eils, R.; et al. Precision oncology based on omics data: The NCT Heidelberg experience. Int. J. Cancer 2017, 141, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Dong, F.; Borger, D.; Dias-Santagata, D.; Ellisen, L.W.; Le, L.P.; Iafrate, A.J. Standardized decision support in next generation sequencing reports of somatic cancer variants. Mol. Oncol 2014, 8, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Palomo, L.; Ibanez, M.; Abaigar, M.; Vazquez, I.; Alvarez, S.; Cabezon, M.; Tazon-Vega, B.; Rapado, I.; Fuster-Tormo, F.; Cervera, J.; et al. Spanish Guidelines for the use of targeted deep sequencing in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hébrant, A.; Punie, K.; Duhoux, F.P.; Colpaert, C.; Floris, G.; Lambein, K.; Neven, P.; Berlière, M.; Salgado, R.; Chintinne, M.; et al. Molecular test algorithms for breast tumours. Belgian J. Med. Oncol. 2019, 13, 40–45. [Google Scholar]
- Hébrant, A.; Jouret-Mourin, A.; Froyen, G.; van der Meulen, J.; de Man, M.; Salgado, R.; van den Eynde, M.; D’haene, N.; Martens, G.; van Cutsem, E.; et al. Molecular test algorithms for digestive tumours. Belgian J. Med. Oncol. 2019, 13, 4–10. [Google Scholar]
- Hébrant, A.; Lammens, M.; Van den Broecke, C.; D’Haene, N.; Van den Oord, J.; Vanderstichele, A.; Dendooven, A.; Neven, P.; Punie, K.; Floris, G.; et al. Algorithms for molecular testing in solid tumors. Belgian J. Med. Oncol. 2019, 13, 286–294. [Google Scholar]
- Van Valckenborgh, E.; Bakkus, M.; Boone, E.; Camboni, A.; Defour, J.-P.; Denys, B.; Devos, H.; Dewispelaere, L.; Froyen, G.; Hébrant, A.; et al. Diagnostic testing in myeloid malignancies by next-generation sequencing: Recommendations from the Commission Personalised Medicine. Belgian J. Hematol. 2019, 10, 241–249. [Google Scholar]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Sukhai, M.A.; Zhang, T.; Dolatshahi, R.; Harbi, D.; Garg, S.; Misyura, M.; Pugh, T.; Stockley, T.L.; Kamel-Reid, S. Integration of Technical, Bioinformatic, and Variant Assessment Approaches in the Validation of a Targeted Next-Generation Sequencing Panel for Myeloid Malignancies. Arch. Pathol. Lab. Med. 2017, 141, 759–775. [Google Scholar] [CrossRef] [PubMed]
- Hébrant, A.; Van Valckenborgh, E.; Salgado, R.; Froyen, G.; Hulstaert, F.; Roberfroid, D.; Tejpar, S.; Jouret-Mourin, A.; Van den Bulcke, M.; Waeytens, A. Opportunities and challenges in oncology and molecular testing: The Belgian strategy. Belgian J. Med. Oncol. 2018, 12, 46–50. [Google Scholar]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Malcikova, J.; Tausch, E.; Rossi, D.; Sutton, L.A.; Soussi, T.; Zenz, T.; Kater, A.P.; Niemann, C.U.; Gonzalez, D.; Davi, F.; et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-update on methodological approaches and results interpretation. Leukemia 2018, 32, 1070–1080. [Google Scholar] [CrossRef]
- Huang, J.; Wang, Y.; Zhai, Y.; Wang, J. Non-small cell lung cancer harboring a rare EGFR L747P mutation showing intrinsic resistance to both gefitinib and osimertinib (AZD9291): A case report. Thorac Cancer 2018, 9, 745–749. [Google Scholar] [CrossRef]
- Wang, Y.T.; Ning, W.W.; Li, J.; Huang, J.A. Exon 19 L747P mutation presented as a primary resistance to EGFR-TKI: A case report. J. Thorac Dis. 2016, 8, E542–E546. [Google Scholar] [CrossRef]
- Mannelli, F.; Ponziani, V.; Bencini, S.; Bonetti, M.I.; Benelli, M.; Cutini, I.; Gianfaldoni, G.; Scappini, B.; Pancani, F.; Piccini, M.; et al. CEBPA-double-mutated acute myeloid leukemia displays a unique phenotypic profile: A reliable screening method and insight into biological features. Haematologica 2017, 102, 529–540. [Google Scholar] [CrossRef]
- Taskesen, E.; Bullinger, L.; Corbacioglu, A.; Sanders, M.A.; Erpelinck, C.A.; Wouters, B.J.; van der Poel-van de Luytgaarde, S.C.; Damm, F.; Krauter, J.; Ganser, A.; et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: Further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 2011, 117, 2469–2475. [Google Scholar] [CrossRef] [PubMed]
- Sukhai, M.A.; Misyura, M.; Thomas, M.; Garg, S.; Zhang, T.; Stickle, N.; Virtanen, C.; Bedard, P.L.; Siu, L.L.; Smets, T.; et al. Somatic Tumor Variant Filtration Strategies to Optimize Tumor-Only Molecular Profiling Using Targeted Next-Generation Sequencing Panels. J. Mol. Diagn. 2018, 2, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Leichsenring, J.; Horak, P.; Kreutzfeldt, S.; Heining, C.; Christopoulos, P.; Volckmar, A.L.; Neumann, O.; Kirchner, M.; Ploeger, C.; Budczies, J.; et al. Variant Classification in Precision Oncology. Int. J. Cancer 2019, 145, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Luo, W.; Wang, M.; Wegman-Ostrosky, T.; Frone, M.N.; Johnston, J.J.; Nickerson, M.L.; Rotunno, M.; Li, S.A.; Achatz, M.I.; et al. Prevalence of pathogenic/likely pathogenic variants in the 24 cancer genes of the ACMG Secondary Findings v2.0 list in a large cancer cohort and ethnicity-matched controls. Genome Med. 2018, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Garber, K.B.; Vincent, L.M.; Alexander, J.J.; Bean, L.J.H.; Bale, S.; Hegde, M. Reassessment of Genomic Sequence Variation to Harmonize Interpretation for Personalized Medicine. Am. J. Hum. Genet. 2016, 99, 1140–1149. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kleinberger, J.; Maloney, K.A.; Pollin, T.I.; Jeng, L.J. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet. Med. 2016, 18, 1165. [Google Scholar] [CrossRef]
- Rivera-Munoz, E.A.; Milko, L.V.; Harrison, S.M.; Azzariti, D.R.; Kurtz, C.L.; Lee, K.; Mester, J.L.; Weaver, M.A.; Currey, E.; Craigen, W.; et al. ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum. Mutat. 2018, 39, 1614–1622. [Google Scholar] [CrossRef]
- Balmana, J.; Digiovanni, L.; Gaddam, P.; Walsh, M.F.; Joseph, V.; Stadler, Z.K.; Nathanson, K.L.; Garber, J.E.; Couch, F.J.; Offit, K.; et al. Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing. J. Clin. Oncol. 2016, 34, 4071–4078. [Google Scholar] [CrossRef]
- Christensen, P.A.; Ni, Y.; Bao, F.; Hendrickson, H.L.; Greenwood, M.; Thomas, J.S.; Long, S.W.; Olsen, R.J. Houston Methodist Variant Viewer: An Application to Support Clinical Laboratory Interpretation of Next-generation Sequencing Data for Cancer. J. Pathol. Inform. 2017, 8, 44. [Google Scholar]
- Lindeboom, R.G.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Harrison, S.M.; Rehm, H.L.; Plon, S.E.; Biesecker, L.G.; ClinGen Sequence Variant Interpretation Working Group. Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum. Mutat. 2018, 39, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- McNulty, S.N.; Parikh, B.A.; Duncavage, E.J.; Heusel, J.W.; Pfeifer, J.D. Optimization of Population Frequency Cut-offs for Filtering Common Germline Polymorphisms from Tumor-Only Next-Generation Sequencing Data. J. Mol. Diagn. 2019, 5, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.T.; Bhattarai, T.S.; Schram, A.M.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; Chakravarty, D.; Phillips, S.; Kandoth, C.; Penson, A.; et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov. 2018, 8, 174–183. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Transcript ID | Hs1 | Hs2 | Hs3 | Hs4 | Hs5 | Hs6 | Hs7 | Hs8 | Hs9 | Hs10 | Hs11 | Hs12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALK | NM_004304.4 | F1174L | R1275Q | ||||||||||
| BRAF | NM_004333.5 | G469A/E/R/V | D594G/M | T599-K601 if-del/ins | V600E/K/M/R | K601E | |||||||
| BRCA1 | NM_007294.3 | all clear LoF variants (nonsense, frameshift, splice site) | |||||||||||
| BRCA2 | NM_000059.3 | all clear LoF variants (nonsense, frameshift, splice site) | |||||||||||
| EGFR | NM_005228.4 | G719A/C/S | ex19if-del/ins | ex20 if-ins | T790M | C797S | L858R | L861Q | |||||
| ESR1 | NM_000125.3 | K303R | E380Q | V392I | S463P | V533M | V534E | P535H | L536H/P/Q/R | Y537C/N/S | D538G | ||
| GNAS | NM_000516.5 | R201C/H | |||||||||||
| H3F3A | NM_002107.4 | K28M | G35R/W | ||||||||||
| HRAS | NM_005343.3 | G12C/D/S/V | G13C/D/R/S/V | Q61H/K/L/R | |||||||||
| IDH1 | NM_005896.3 | R132C/G/H/L/S | |||||||||||
| IDH2 | NM_002168.3 | R140L/Q/W | R172K/M/S | ||||||||||
| KIT | NM_000222.2 | ex8 | ex9 | ex11 | ex11 | ex11 | ex11 | ex11 | ex13 | ex13 | ex14 | ex17 | ex17 |
| D419 if-del | S501-F504 if-ins | K550-V560 if-indel | W557G/R | V559A/D | V560D | L576P | K642E | V654A | T670I | D816H/V/Y | N822K | ||
| KRAS | NM_004985.4 | G12A/C/D/F/R/S/V | G13C/D/R/S/V | A59T | Q61H/K/L/R | K117N | A146T | ||||||
| MET | NM_001127500.3 | ex14 skipping | |||||||||||
| NRAS | NM_002524.4 | G12A/C/D/R/S/V | G13C/D/R/S/V | A59T | Q61H/K/L/R | K117N | A146T | ||||||
| PDGFRA | NM_006206.5 | S566_E577 if-del | D842V | D842_I843 if-del | V561D | ||||||||
| Gene | Transcript ID | Hs1 | Hs2 | Hs3 | Hs4 | Hs5 | Hs6 |
|---|---|---|---|---|---|---|---|
| ASXL1 | NM_015338.5 | none | |||||
| CALR | NM_004343.3 | ex9of-del | ex9of-ins | ||||
| CEBPA | NM_004364.3 | none | |||||
| CSF3R | NM_156039.3 | T618I | |||||
| DNMT3A | NM_ 175629.2 | R882C/H | |||||
| EZH2 | NM_004456.4 | Y646F/H/N/S | |||||
| FLT3 | NM_004119.2 | ex14if-dup | D835A/E/H/V/Y | ||||
| IDH1 | NM_005896.3 | R132C/G/H/L/S | |||||
| IDH2 | NM_002168.3 | R140L/Q/W | R172K/M/S | ||||
| JAK2 | NM_004972.3 | ex12 if-del/if-dup | V617F | ||||
| KIT | NM_000222.2 | see CPV Solid list | |||||
| MPL | NM_005373.2 | S505N | W515any ms | ||||
| NPM1 | NM_002520.6 | ex11of-ins | |||||
| RUNX1 | NM_001754.4 | none | |||||
| SETBP1 | NM_015559.3 | D868N | G870S | ||||
| SF3B1 | NM_012433.3 | E622D | R625C/H | H662Q | K666N/R/T | K700E | G742D |
| SRSF2 | NM_003016.4 | P95H/L/R | P95_R102del | ||||
| TET2 | NM_001127208.2 | none | |||||
| TP53 | NM_000546.5 | R175H | Y220C | G245S | R248Q/W | R273C/H | R282W |
| U2AF1 | NM_006758.2 | S34F/Y | Q157P/R | ||||
| WT1 | NM_024426.5 | none |
| Parameter | Score +2 | Score +1 | Score +0.5 | Score 0 | Score −1 |
|---|---|---|---|---|---|
| Total # of entries of that particular AA change at that position in COSMIC | Solid: ≥50 | 50 > x > 10 | / | ≤10 | / |
| Hemato: ≥10 | 10 > x > 5 | / | ≤5 | / | |
| In silico prediction tools SIFT and MutationTaster | / | / | Both damaging and deleterious | Other | / |
| Harmful in functional studies (PubMed, JAX-CKB, MDA, MCG) | / | / | Yes | Not reported | No |
| Described in at least one genomic db (CIVIC, ClinVar, OncoKb, VarSome) | / | / | As (Likely) Pathogenic | Not described/unknown | As (Likely) Benign |
| Parameter | Example(s) |
|---|---|
| Sample ID (primary lab) | 123-45678 |
| Sampling date | 16th January 2019 |
| Date of sample received | 17th January 2019 |
| Sample tumoral stage | primary, metastasis |
| Sample anatomic site | colon, liver, blood, lymph node, … |
| Sample type | resection, (trephine) biopsy, aspirate, … |
| Sample procedure | FFPE, fresh frozen, fresh tissue, … |
| Neoplastic cells (%) | 30%, na |
| Sample quality | disclaimer if sample does not fulfill pre-analytical requirements |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Froyen, G.; Le Mercier, M.; Lierman, E.; Vandepoele, K.; Nollet, F.; Boone, E.; Van der Meulen, J.; Jacobs, K.; Lambin, S.; Vander Borght, S.; et al. Standardization of Somatic Variant Classifications in Solid and Haematological Tumours by a Two-Level Approach of Biological and Clinical Classes: An Initiative of the Belgian ComPerMed Expert Panel. Cancers 2019, 11, 2030. https://doi.org/10.3390/cancers11122030
Froyen G, Le Mercier M, Lierman E, Vandepoele K, Nollet F, Boone E, Van der Meulen J, Jacobs K, Lambin S, Vander Borght S, et al. Standardization of Somatic Variant Classifications in Solid and Haematological Tumours by a Two-Level Approach of Biological and Clinical Classes: An Initiative of the Belgian ComPerMed Expert Panel. Cancers. 2019; 11(12):2030. https://doi.org/10.3390/cancers11122030
Chicago/Turabian StyleFroyen, Guy, Marie Le Mercier, Els Lierman, Karl Vandepoele, Friedel Nollet, Elke Boone, Joni Van der Meulen, Koen Jacobs, Suzan Lambin, Sara Vander Borght, and et al. 2019. "Standardization of Somatic Variant Classifications in Solid and Haematological Tumours by a Two-Level Approach of Biological and Clinical Classes: An Initiative of the Belgian ComPerMed Expert Panel" Cancers 11, no. 12: 2030. https://doi.org/10.3390/cancers11122030
APA StyleFroyen, G., Le Mercier, M., Lierman, E., Vandepoele, K., Nollet, F., Boone, E., Van der Meulen, J., Jacobs, K., Lambin, S., Vander Borght, S., Van Valckenborgh, E., Antoniou, A., & Hébrant, A. (2019). Standardization of Somatic Variant Classifications in Solid and Haematological Tumours by a Two-Level Approach of Biological and Clinical Classes: An Initiative of the Belgian ComPerMed Expert Panel. Cancers, 11(12), 2030. https://doi.org/10.3390/cancers11122030