Inhibition of Histone Demethylases LSD1 and UTX Regulates ERα Signaling in Breast Cancer

 ,
,  , ,
, ,  , ,
, ,  , , , and
, , , and  add
Show full author list
add
Show full author list
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
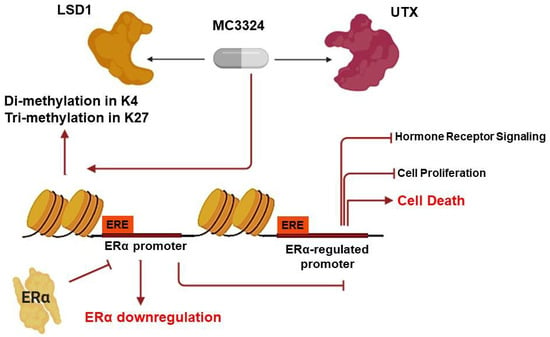
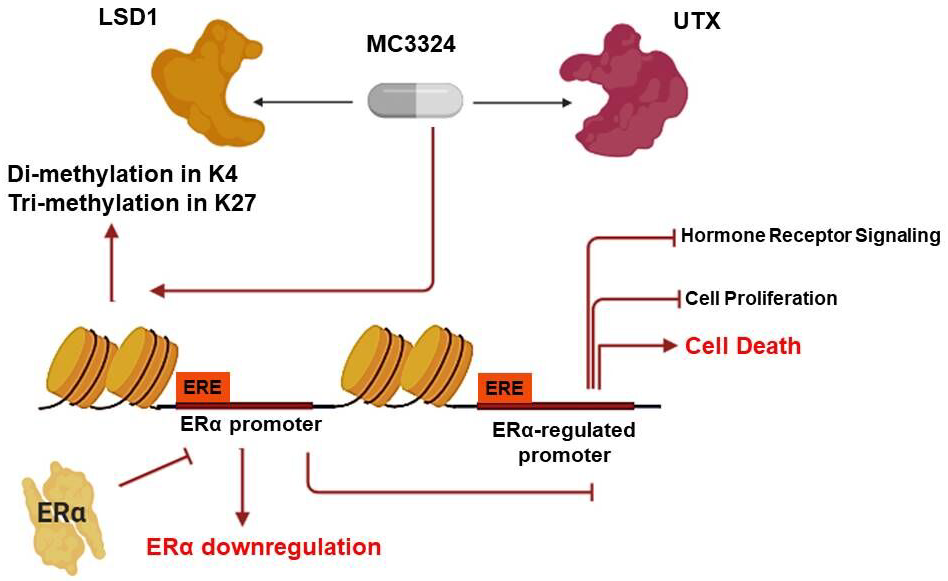
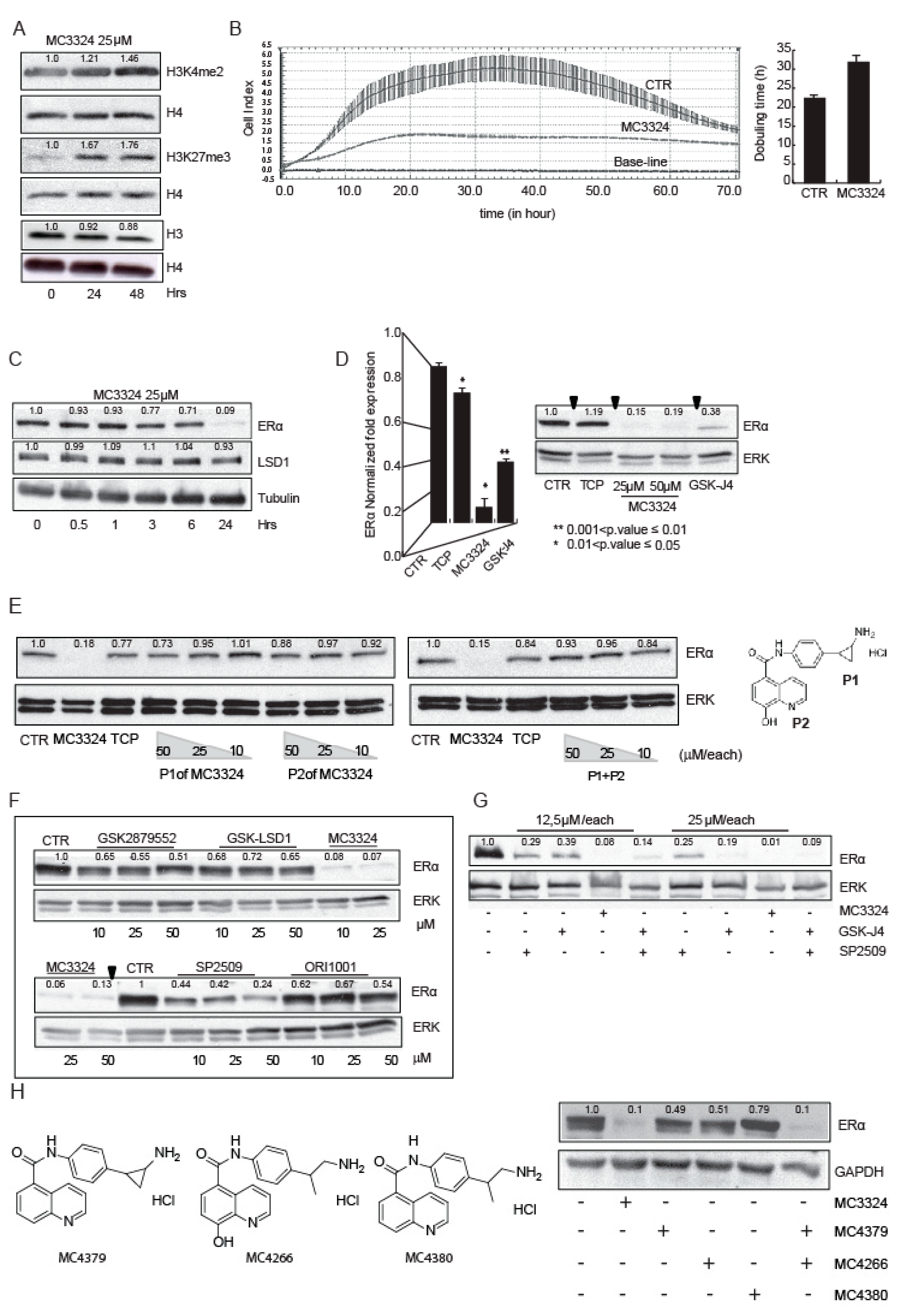
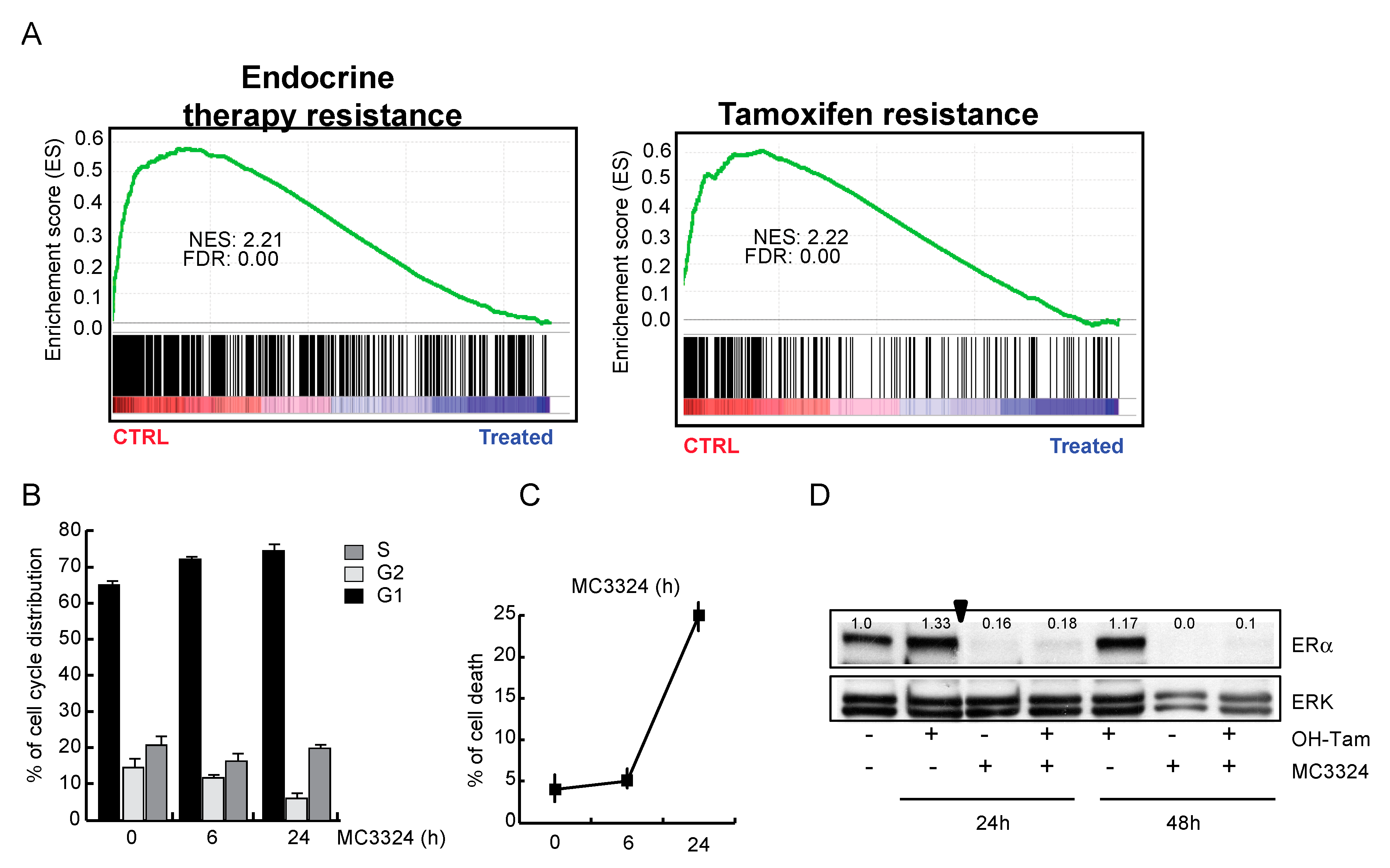
2.1. MC3324 Is a Dual LSD1 and UTX Inhibitor Regulating ERα Signaling
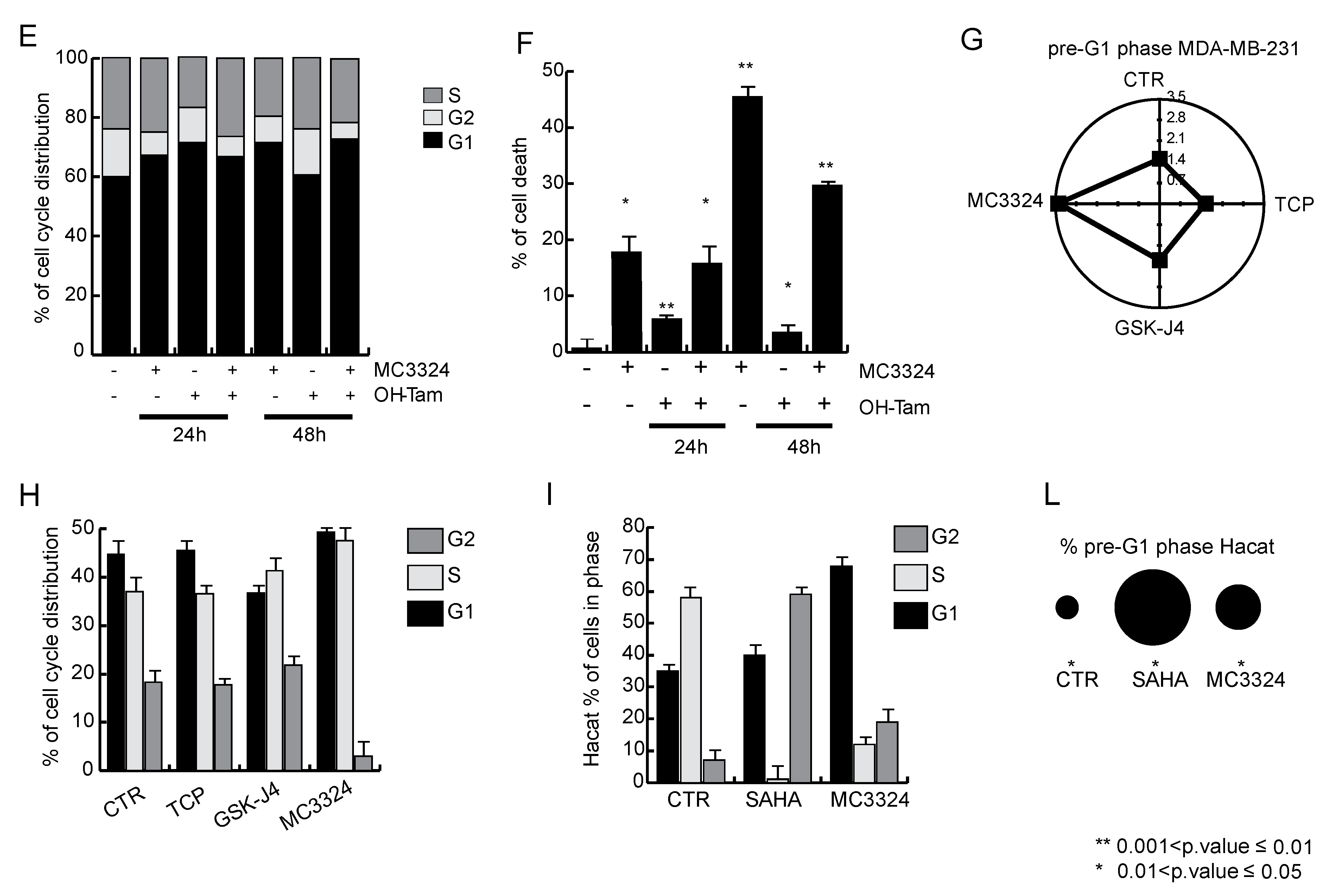
2.2. MC3324 Blocks Proliferation of Tamoxifen-Insensitive BC Cell Line
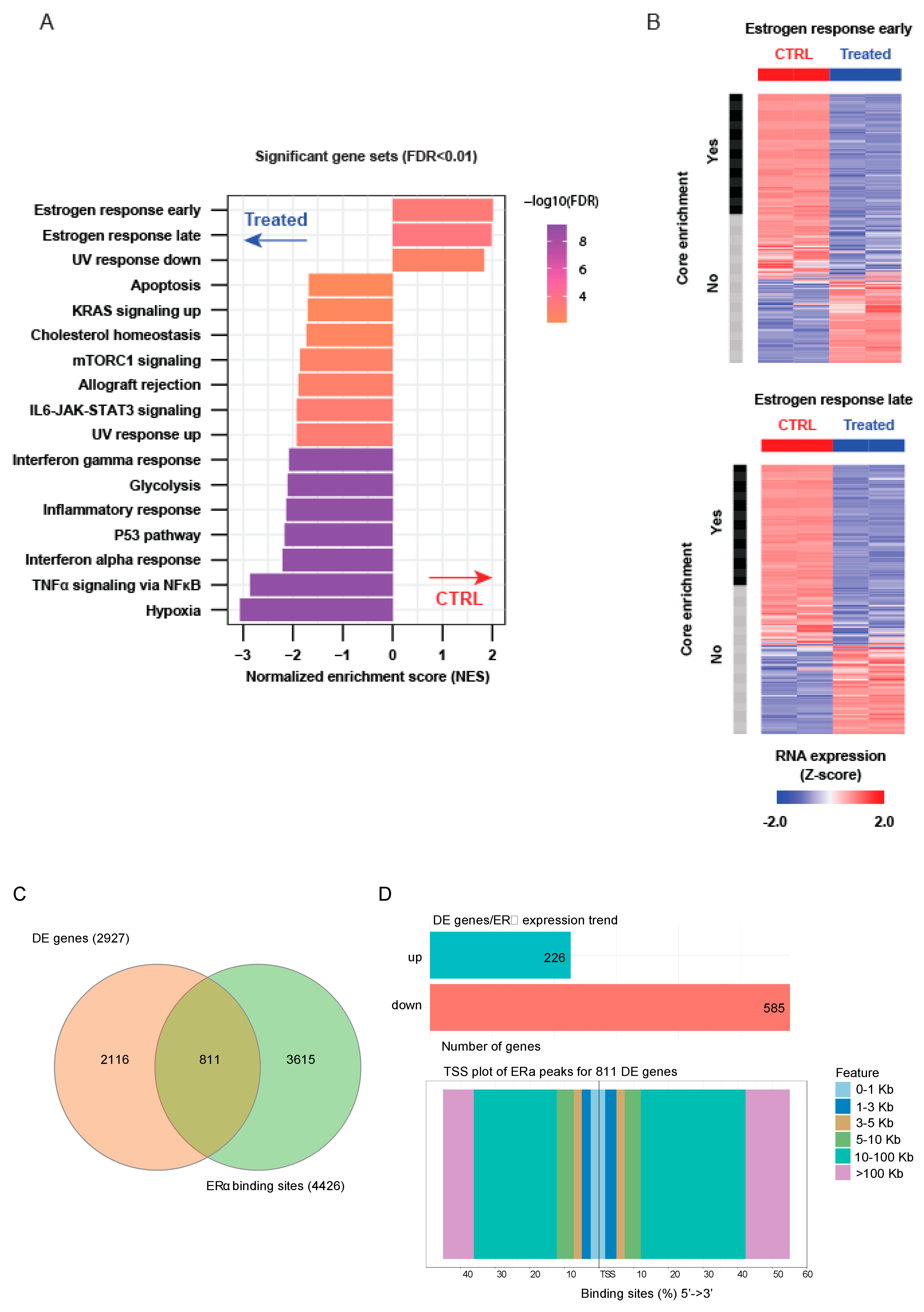
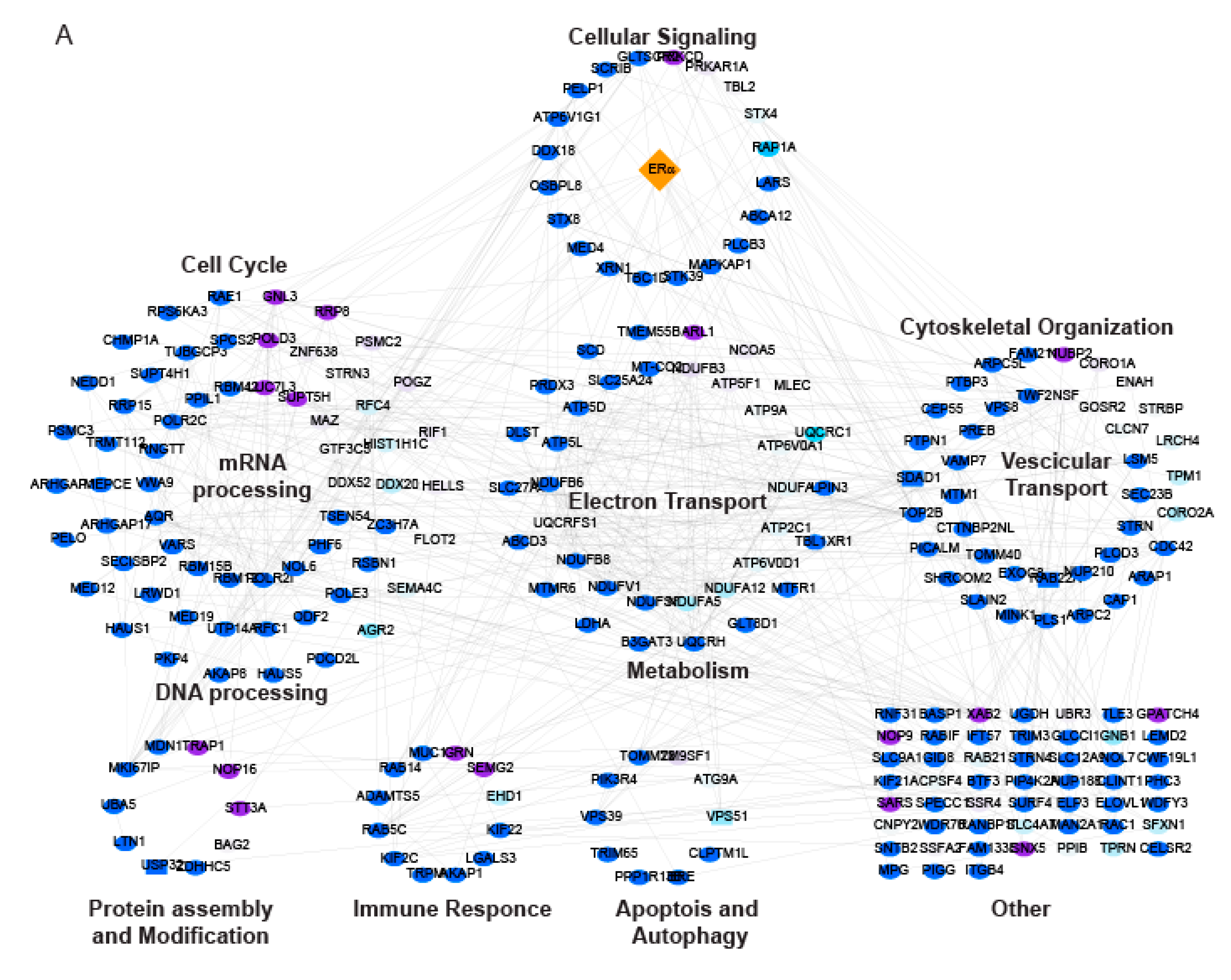
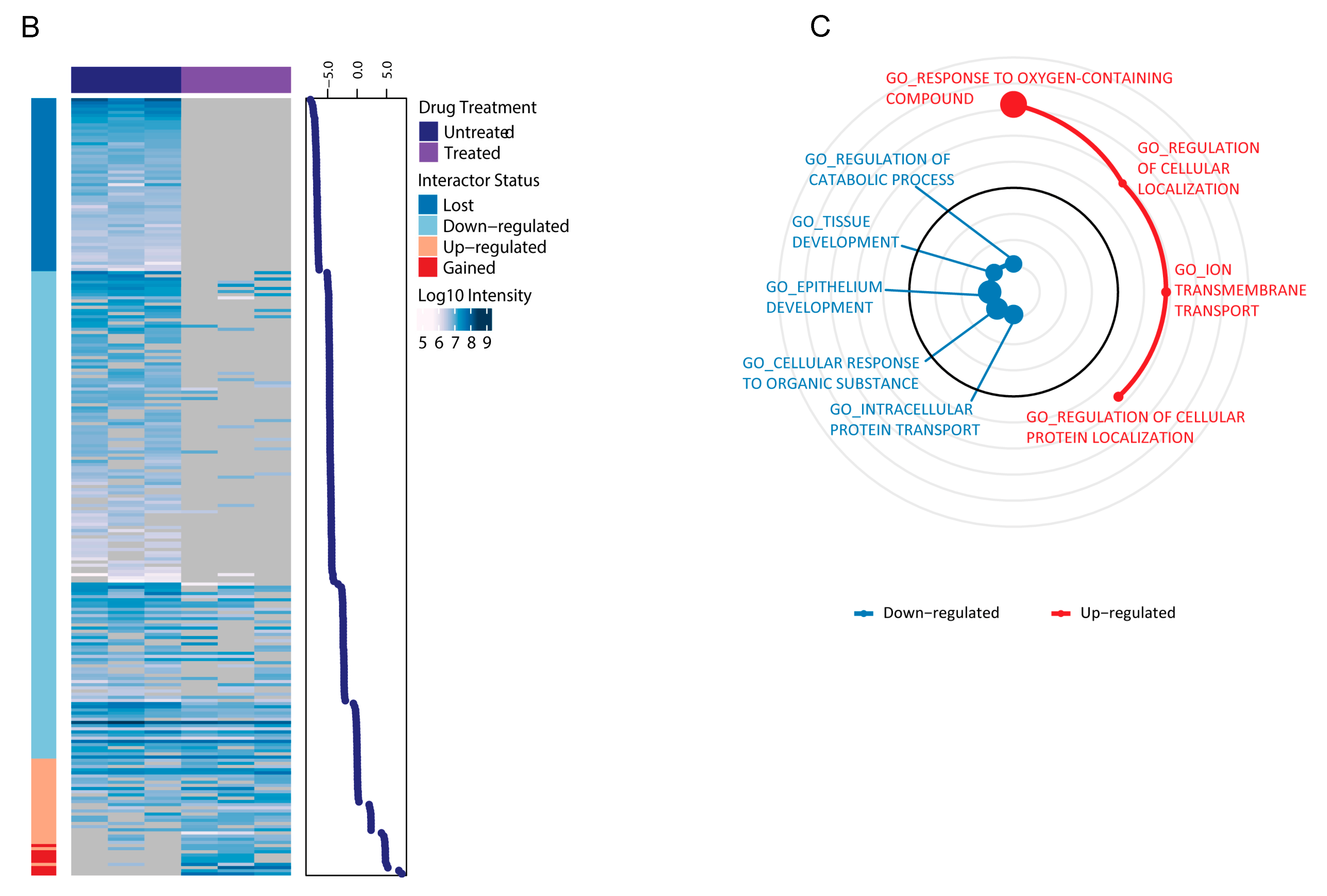
2.3. LSD1 and UTX Inhibition Modulates ERα Interactome and Hormone Signaling Cascade
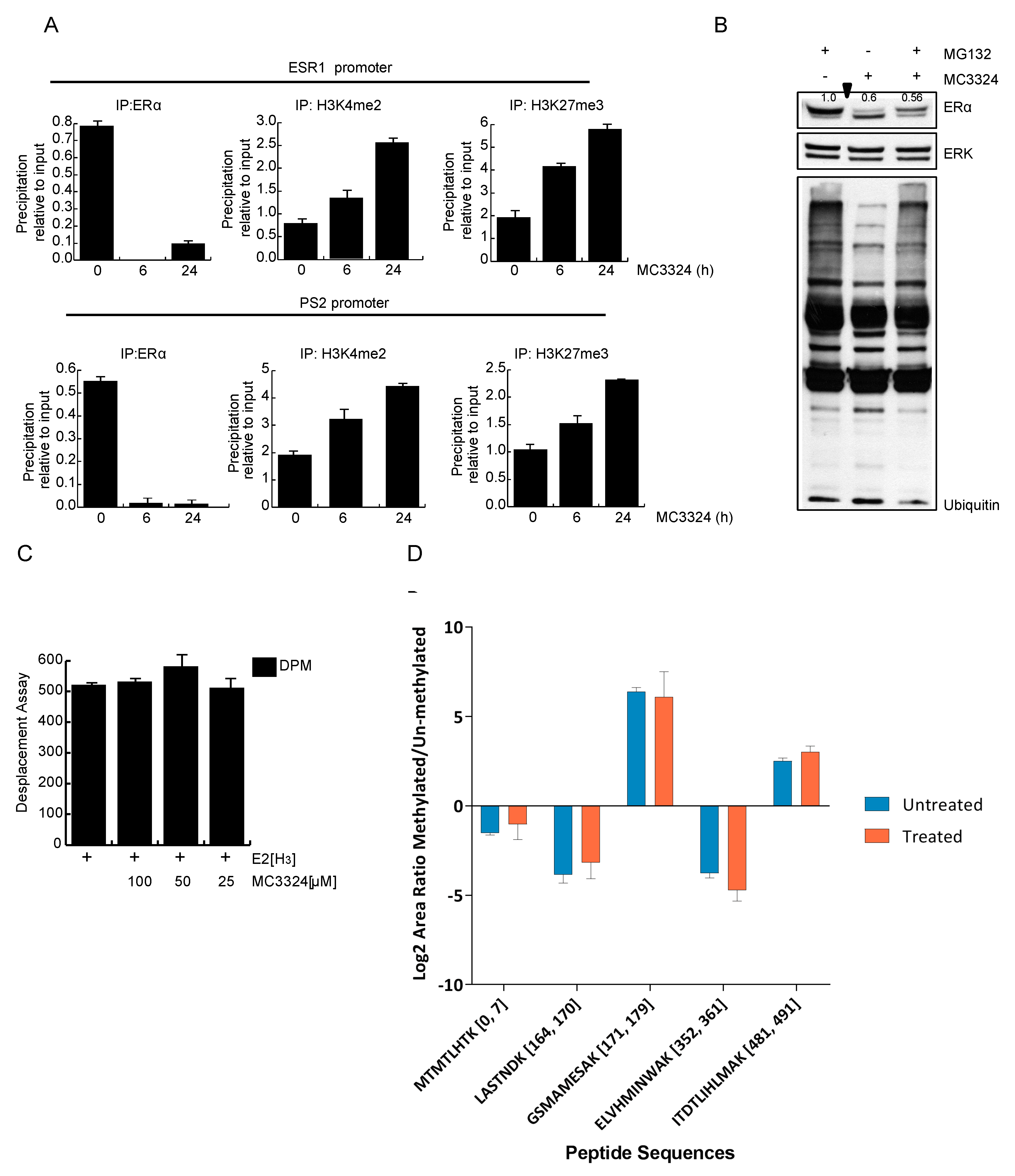
2.4. Epigenetic Rebalance of Erα Signaling via LSD1 and UTX Inhibition
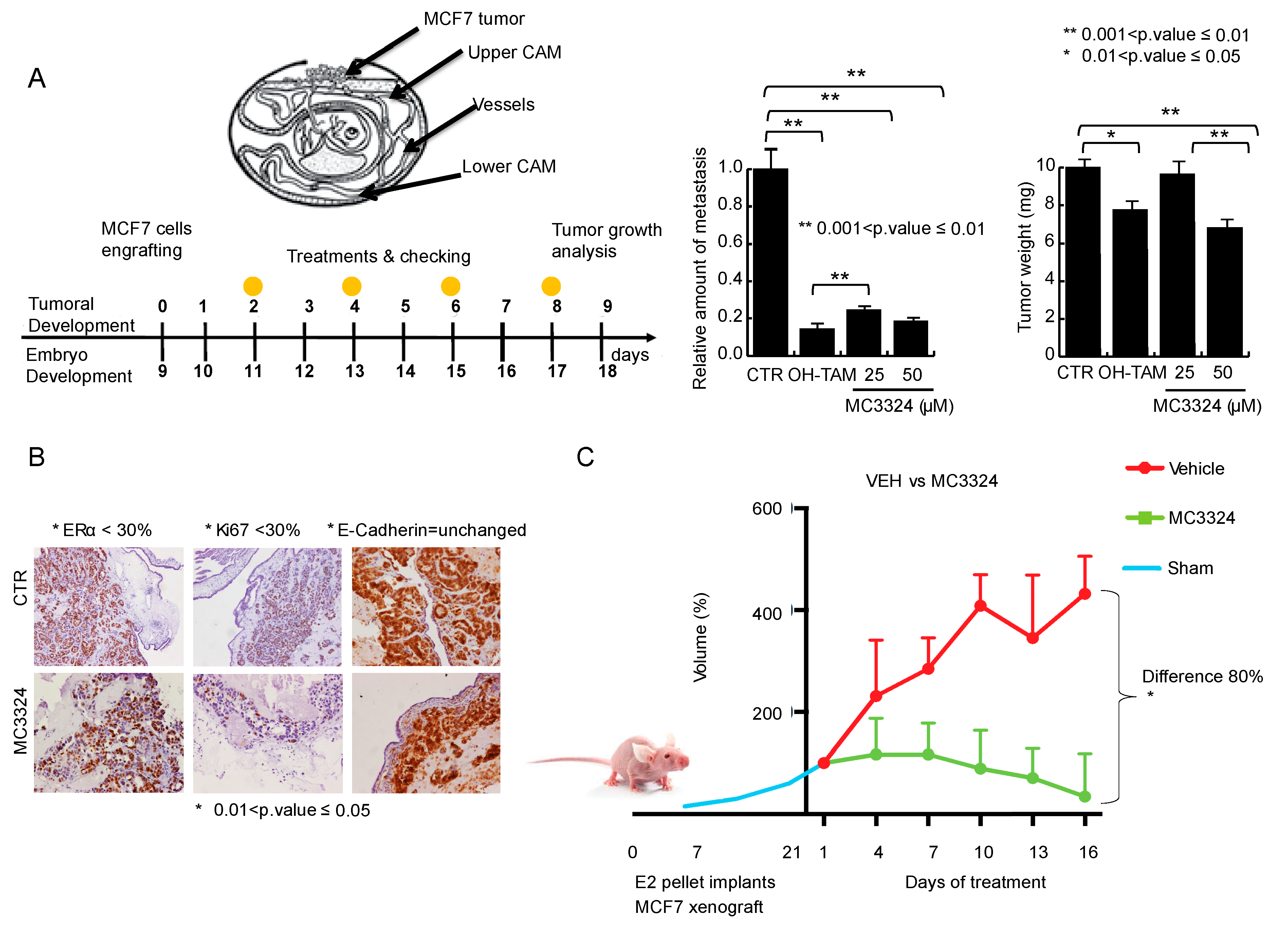
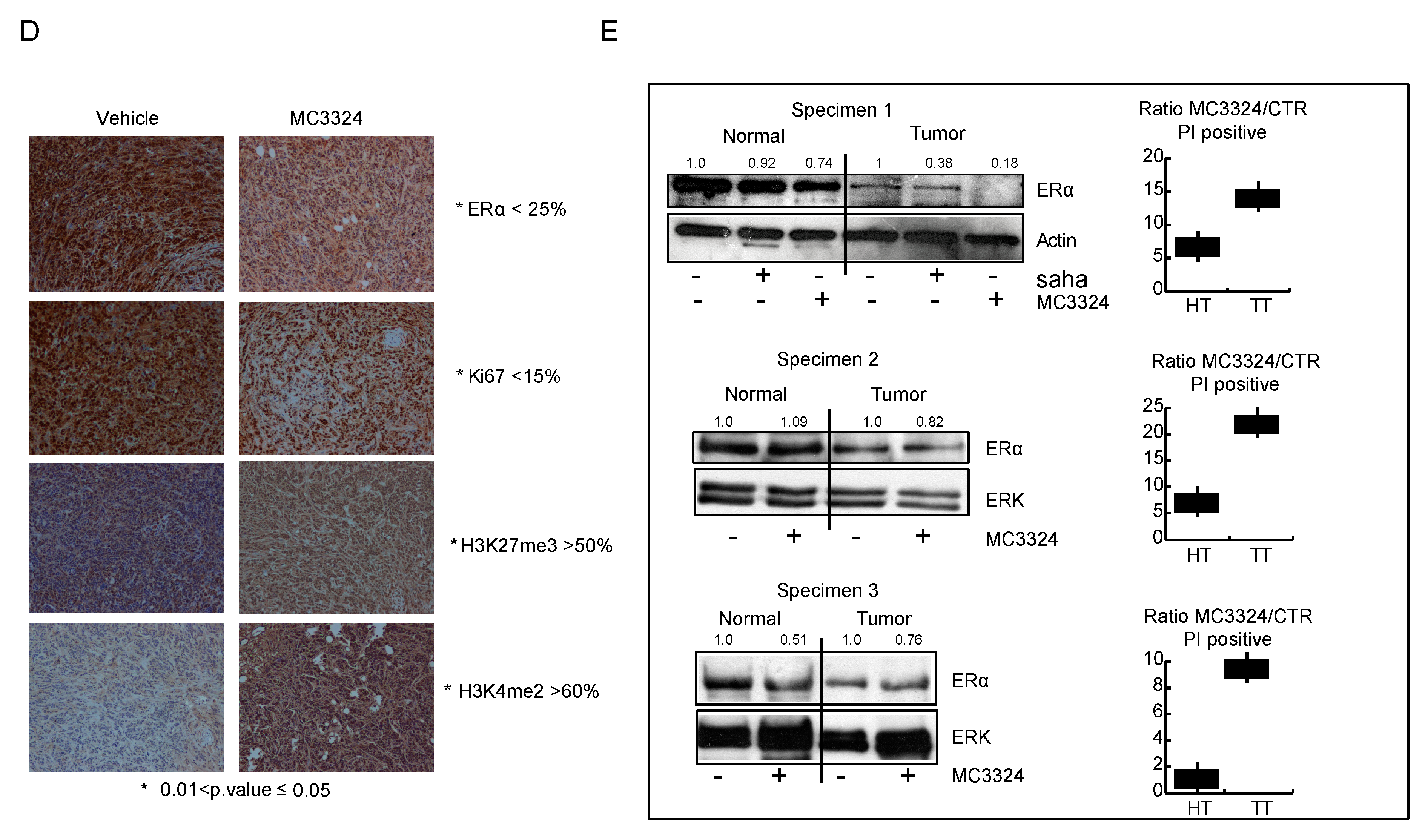
2.5. MC3324 Displays Anticancer Action In Vivo in Both Chicken Embryo and Mouse Models and Ex Vivo in Human BC Specimens
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Cycle
4.4. Histone Extraction
4.5. Western Blot Analysis
4.6. RNA Isolation and Quantitation
4.7. Cellular Thermal Shift Assay(CETSA)
4.8. Cell Proliferation Assay
4.9. RNA-Seq and Statistical Analysis
4.10. Co-Immunoprecipitation (Co-IP) and Mass Spectrometry (MS) Analysis
4.11. ERα-E2 Radiolabeled Displacement Assay
4.12. Isolation of Cells from Ex Vivo Biopsies
4.13. Chromatin Immunoprecipitation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Saad, E.D.; Barrios, C.H.; Bines, J. Clinical implications of ESR1 mutations in hormone receptor-positive advanced breast cancer. Front. Oncol. 2017, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M.; Cuzick, J.; Ingle, J.; Coates, A.; Forbes, J.; Bliss, J.; Buyse, M.; Baum, M.; Buzdar, A.; Colleoni, M.; et al. Meta-analysis of breast cancer outcomes in adjuvant trials of aromatase inhibitors versus tamoxifen. J. Clin. Oncol. 2010, 28, 509–518. [Google Scholar] [CrossRef]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. (Seoul) 2012, 20, 256–267. [Google Scholar] [CrossRef]
- Howell, A. Pure oestrogen antagonists for the treatment of advanced breast cancer. Endocr. Relat. Cancer 2006, 13, 689–706. [Google Scholar] [CrossRef]
- Nathan, M.R.; Schmid, P. A review of fulvestrant in breast cancer. Oncol. Ther. 2017, 5, 17–29. [Google Scholar] [CrossRef]
- Zhang, C.; Guo, S.; Yang, L.; Liu, J.; Zheng, S.; Zhong, Q.; Zhang, Q.; Wang, G. Metabolism, pharmacokinetics, and bioavailability of ZB716, a Steroidal Selective Estrogen Receptor Downregulator (SERD). Oncotarget 2017, 8, 103874–103889. [Google Scholar] [CrossRef]
- Boer, K. Fulvestrant in advanced breast cancer: evidence to date and place in therapy. Ther. Adv. Med. Oncol. 2017, 9, 465–479. [Google Scholar] [CrossRef]
- Yang, J.; Jubb, A.M.; Pike, L.; Buffa, F.M.; Turley, H.; Baban, D.; Leek, R.; Gatter, K.C.; Ragoussis, J.; Harris, A.L. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010, 70, 6456–6466. [Google Scholar] [CrossRef] [PubMed]
- Ombra, M.N.; Di Santi, A.; Abbondanza, C.; Migliaccio, A.; Avvedimento, E.V.; Perillo, B. Retinoic acid impairs estrogen signaling in breast cancer cells by interfering with activation of LSD1 via PKA. Biochim. Biophys. Acta 2013, 1829, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 2010, 29, 3691–3702. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Sphyris, N.; Johnson, K.S.; Reisenauer, K.N.; Nesbit, T.A.; Joseph, R.; Vijay, G.V.; Sarkar, T.R.; Bhangre, N.A.; Song, J.J.; et al. The H3K27me3-demethylase KDM6A is suppressed in breast cancer stem-like cells, and enables the resolution of bivalency during the mesenchymal-epithelial transition. Oncotarget 2017, 8, 65548–65565. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Lee, S.H.; Chen, K.; Zhu, G.; Oh, W.; Allton, K.; Gafni, O.; Kim, Y.Z.; Tomoiga, A.S.; Barton, M.C.; et al. An essential role for UTX in resolution and activation of bivalent promoters. Nucleic Acids Res. 2016, 44, 3659–3674. [Google Scholar] [CrossRef]
- Xie, G.; Liu, X.; Zhang, Y.; Li, W.; Liu, S.; Chen, Z.; Xu, B.; Yang, J.; He, L.; Zhang, Z.; et al. UTX promotes hormonally responsive breast carcinogenesis through feed-forward transcription regulation with estrogen receptor. Oncogene 2017, 36, 5497–5511. [Google Scholar] [CrossRef]
- Rotili, D.; Tomassi, S.; Conte, M.; Benedetti, R.; Tortorici, M.; Ciossani, G.; Valente, S.; Marrocco, B.; Labella, D.; Novellino, E.; et al. Pan-histone demethylase inhibitors simultaneously targeting Jumonji C and lysine-specific demethylases display high anticancer activities. J. Med. Chem. 2014, 57, 42–55. [Google Scholar] [CrossRef]
- Thinnes, C.C.; England, K.S.; Kawamura, A.; Chowdhury, R.; Schofield, C.J.; Hopkinson, R.J. Targeting histone lysine demethylases - progress, challenges, and the future. Biochim. Biophys. Acta 2014, 1839, 1416–1432. [Google Scholar] [CrossRef]
- Rajabi, H.; Kufe, D. MUC1-C oncoprotein integrates a program of emt, epigenetic reprogramming and immune evasion in human carcinomas. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 117–122. [Google Scholar] [CrossRef]
- Wei, X.; Xu, H.; Kufe, D. MUC1 oncoprotein stabilizes and activates estrogen receptor alpha. Mol. Cell 2006, 21, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.Z.; Barnea, I.; Aylon, Y.; Gorivodsky, M.; Wreschner, D.H.; Keydar, I. MUC1 gene overexpressed in breast cancer: structure and transcriptional activity of the MUC1 promoter and role of estrogen receptor alpha (ERalpha) in regulation of the MUC1 gene expression. Mol. Cancer 2006, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Finucane, H.K.; Schumacher, F.R.; Schmit, S.L.; Tyrer, J.P.; Han, Y.; Michailidou, K.; Lesseur, C.; Kuchenbaecker, K.B.; Dennis, J.; et al. Shared heritability and functional enrichment across six solid cancers. Nat. Commun. 2019, 10, 431. [Google Scholar] [CrossRef] [PubMed]
- Jabour, A.M.; Dixon, B.E. Monitoring public health reporting: data tracking in cancer registries. Online J. Public Health Inform. 2018, 10, e220. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Effects of tamoxifen in relation to breast cancer. Br. Med. J. 1977, 1, 1534–1535. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, J.; Li, J.; Wang, H.; Wang, Y.; He, Q.; Xia, X.; Hu, Z.Y.; Ouyang, Q. Clinical and genetic risk factors for Fulvestrant treatment in post-menopause ER-positive advanced breast cancer patients. J. Transl. Med. 2019, 17, 27. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Meegan, M.J. Designed multiple ligands for cancer therapy. Curr. Med. Chem. 2011, 18, 4722–4737. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef]
- Park, U.H.; Kang, M.R.; Kim, E.J.; Kwon, Y.S.; Hur, W.; Yoon, S.K.; Song, B.J.; Park, J.H.; Hwang, J.T.; Jeong, J.C.; et al. ASXL2 promotes proliferation of breast cancer cells by linking ERalpha to histone methylation. Oncogene 2016, 35, 3742–3752. [Google Scholar] [CrossRef]
- Bae, W.K.; Yoo, K.H.; Lee, J.S.; Kim, Y.; Chung, I.J.; Park, M.H.; Yoon, J.H.; Furth, P.A.; Hennighausen, L. The methyltransferase EZH2 is not required for mammary cancer development, although high EZH2 and low H3K27me3 correlate with poor prognosis of ER-positive breast cancers. Mol. Carcinog. 2015, 54, 1172–1180. [Google Scholar] [CrossRef]
- Kim, J.H.; Sharma, A.; Dhar, S.S.; Lee, S.H.; Gu, B.; Chan, C.H.; Lin, H.K.; Lee, M.G. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014, 74, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Schulz, W.A.; Lang, A.; Koch, J.; Greife, A. The histone demethylase UTX/KDM6A in cancer: Progress and puzzles. Int. J. Cancer 2019, 145, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Magliulo, D.; Bernardi, R.; Messina, S. Lysine-Specific Demethylase 1A as a promising target in acute myeloid leukemia. Front. Oncol. 2018, 8, 255. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Konopleva, M.Y. ORY-1001: overcoming the differentiation block in AML. Cancer Cell 2018, 33, 342–343. [Google Scholar] [CrossRef]
- Maes, T.; Mascaro, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 2018, 33, 495–511. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, M.; Sheng, M.; Zhang, P.; Chen, Z.; Xing, W.; Bai, J.; Cheng, T.; Yang, F.C.; Zhou, Y. Therapeutic potential of GSK-J4, a histone demethylase KDM6B/JMJD3 inhibitor, for acute myeloid leukemia. J Cancer Res Clin Oncol 2018, 144, 1065–1077. [Google Scholar] [CrossRef]
- Park, J.W.; Cho, H.; Oh, H.; Kim, J.Y.; Seo, S.B. AURKA suppresses leukemic THP-1 cell differentiation through inhibition of the KDM6B pathway. Mol. Cells 2018, 41, 444–453. [Google Scholar] [CrossRef]
- Morozov, V.M.; Li, Y.; Clowers, M.M.; Ishov, A.M. Inhibitor of H3K27 demethylase JMJD3/UTX GSK-J4 is a potential therapeutic option for castration resistant prostate cancer. Oncotarget 2017, 8, 62131–62142. [Google Scholar] [CrossRef]
- Bennesch, M.A.; Segala, G.; Wider, D.; Picard, D. LSD1 engages a corepressor complex for the activation of the estrogen receptor alpha by estrogen and cAMP. Nucleic Acids Res. 2016, 44, 8655–8670. [Google Scholar] [CrossRef]
- Kim, J.; Park, U.H.; Moon, M.; Um, S.J.; Kim, E.J. Negative regulation of ERalpha by a novel protein CAC1 through association with histone demethylase LSD1. FEBS Lett. 2013, 587, 17–22. [Google Scholar] [CrossRef]
- Li, W.; Xu, L.; Che, X.; Li, H.; Zhang, Y.; Song, N.; Wen, T.; Hou, K.; Yang, Y.; Zhou, L.; et al. C-Cbl reverses HER2-mediated tamoxifen resistance in human breast cancer cells. BMC Cancer 2018, 18, 507. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, Y.; Kane, S.E.; Chen, S. Improvement of sensitivity to tamoxifen in estrogen receptor-positive and Herceptin-resistant breast cancer cells. J. Mol. Endocrinol. 2008, 41, 367–377. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaur, J.; Singh, M.; Dell’Aversana, C.; Benedetti, R.; Giardina, P.; Rossi, M.; Valadan, M.; Vergara, A.; Cutarelli, A.; Montone, A.M.I.; et al. Biological interactions of biocompatible and water-dispersed MoS2 nanosheets with bacteria and human cells. Sci. Rep. 2018, 8, 16386. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Nebbioso, A.; Cuomo, F.; Rotili, D.; Cobellis, G.; Bontempo, P.; Baldi, A.; Spugnini, E.P.; Citro, G.; Chambery, A.; et al. RIP1-HAT1-SIRT complex identification and targeting in treatment and prevention of cancer. Clin. Cancer Res. 2018, 24, 2886–2900. [Google Scholar] [CrossRef]
- Conte, M.; Dell’Aversana, C.; Benedetti, R.; Petraglia, F.; Carissimo, A.; Petrizzi, V.B.; D’Arco, A.M.; Abbondanza, C.; Nebbioso, A.; Altucci, L. HDAC2 deregulation in tumorigenesis is causally connected to repression of immune modulation and defense escape. Oncotarget 2015, 6, 886–901. [Google Scholar] [CrossRef]
- Dell’Aversana, C.; Giorgio, C.; D’Amato, L.; Lania, G.; Matarese, F.; Saeed, S.; Di Costanzo, A.; Belsito Petrizzi, V.; Ingenito, C.; Martens, J.H.A.; et al. miR-194-5p/BCLAF1 deregulation in AML tumorigenesis. Leukemia 2017, 31, 2315–2325. [Google Scholar] [CrossRef]
- Franci, G.; Sarno, F.; Nebbioso, A.; Altucci, L. Identification and characterization of PKF118-310 as a KDM4A inhibitor. Epigenetics 2017, 12, 198–205. [Google Scholar] [CrossRef]
- Denissov, S.; van Driel, M.; Voit, R.; Hekkelman, M.; Hulsen, T.; Hernandez, N.; Grummt, I.; Wehrens, R.; Stunnenberg, H. Identification of novel functional TBP-binding sites and general factor repertoires. EMBO J. 2007, 26, 944–954. [Google Scholar] [CrossRef]
- Saeed, S.; Logie, C.; Francoijs, K.J.; Frigè, G.; Romanenghi, M.; Nielsen, F.G.; Raats, L.; Shahhoseini, M.; Huynen, M.; Altucci, L.; et al. Chromatin accessibility, p300, and histone acetylation define PML-RARα and AML1-ETO binding sites in acute myeloid leukemia. Blood 2012, 120(15), 3058–3068. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benedetti, R.; Dell’Aversana, C.; De Marchi, T.; Rotili, D.; Liu, N.Q.; Novakovic, B.; Boccella, S.; Di Maro, S.; Cosconati, S.; Baldi, A.; et al. Inhibition of Histone Demethylases LSD1 and UTX Regulates ERα Signaling in Breast Cancer. Cancers 2019, 11, 2027. https://doi.org/10.3390/cancers11122027
Benedetti R, Dell’Aversana C, De Marchi T, Rotili D, Liu NQ, Novakovic B, Boccella S, Di Maro S, Cosconati S, Baldi A, et al. Inhibition of Histone Demethylases LSD1 and UTX Regulates ERα Signaling in Breast Cancer. Cancers. 2019; 11(12):2027. https://doi.org/10.3390/cancers11122027
Chicago/Turabian StyleBenedetti, Rosaria, Carmela Dell’Aversana, Tommaso De Marchi, Dante Rotili, Ning Qing Liu, Boris Novakovic, Serena Boccella, Salvatore Di Maro, Sandro Cosconati, Alfonso Baldi, and et al. 2019. "Inhibition of Histone Demethylases LSD1 and UTX Regulates ERα Signaling in Breast Cancer" Cancers 11, no. 12: 2027. https://doi.org/10.3390/cancers11122027
APA StyleBenedetti, R., Dell’Aversana, C., De Marchi, T., Rotili, D., Liu, N. Q., Novakovic, B., Boccella, S., Di Maro, S., Cosconati, S., Baldi, A., Niméus, E., Schultz, J., Höglund, U., Maione, S., Papulino, C., Chianese, U., Iovino, F., Federico, A., Mai, A., ... Altucci, L. (2019). Inhibition of Histone Demethylases LSD1 and UTX Regulates ERα Signaling in Breast Cancer. Cancers, 11(12), 2027. https://doi.org/10.3390/cancers11122027