p53-Mediated Tumor Suppression: DNA-Damage Response and Alternative Mechanisms

Abstract
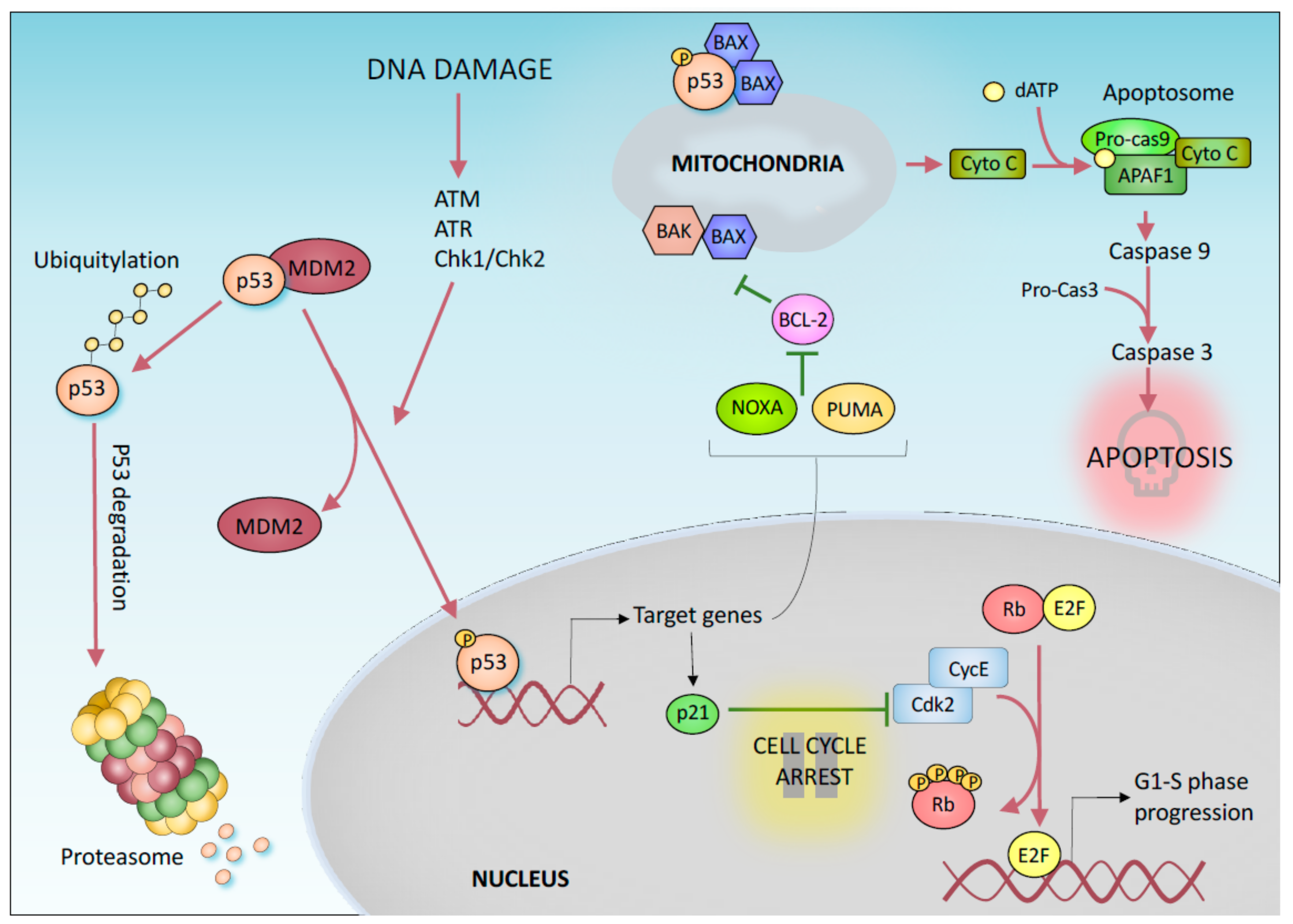
1. “Canonical” p53-Mediated Tumor Suppression
2. “Non-Canonical” p53-Mediated Tumor Suppression
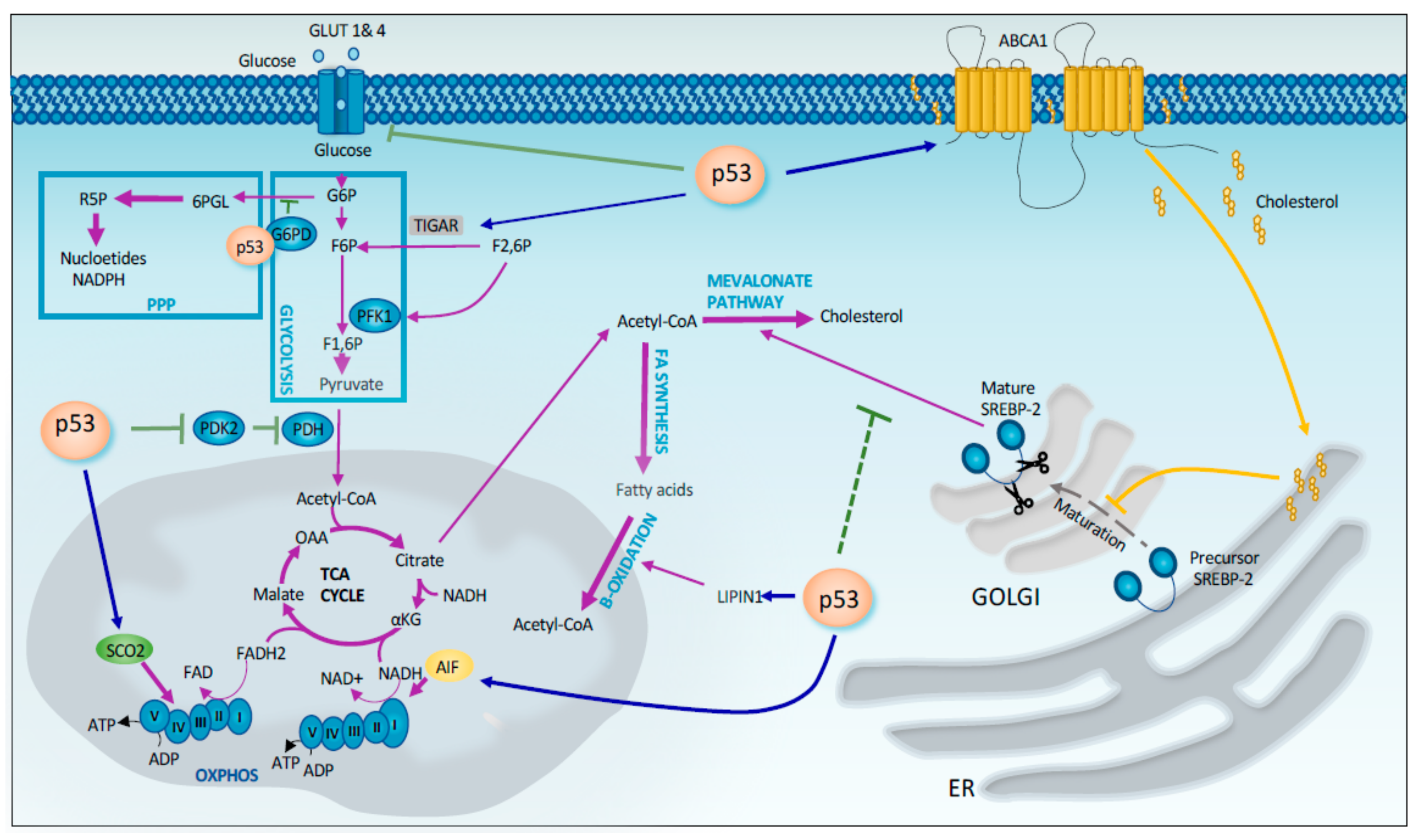
2.1. Regulation of Metabolism
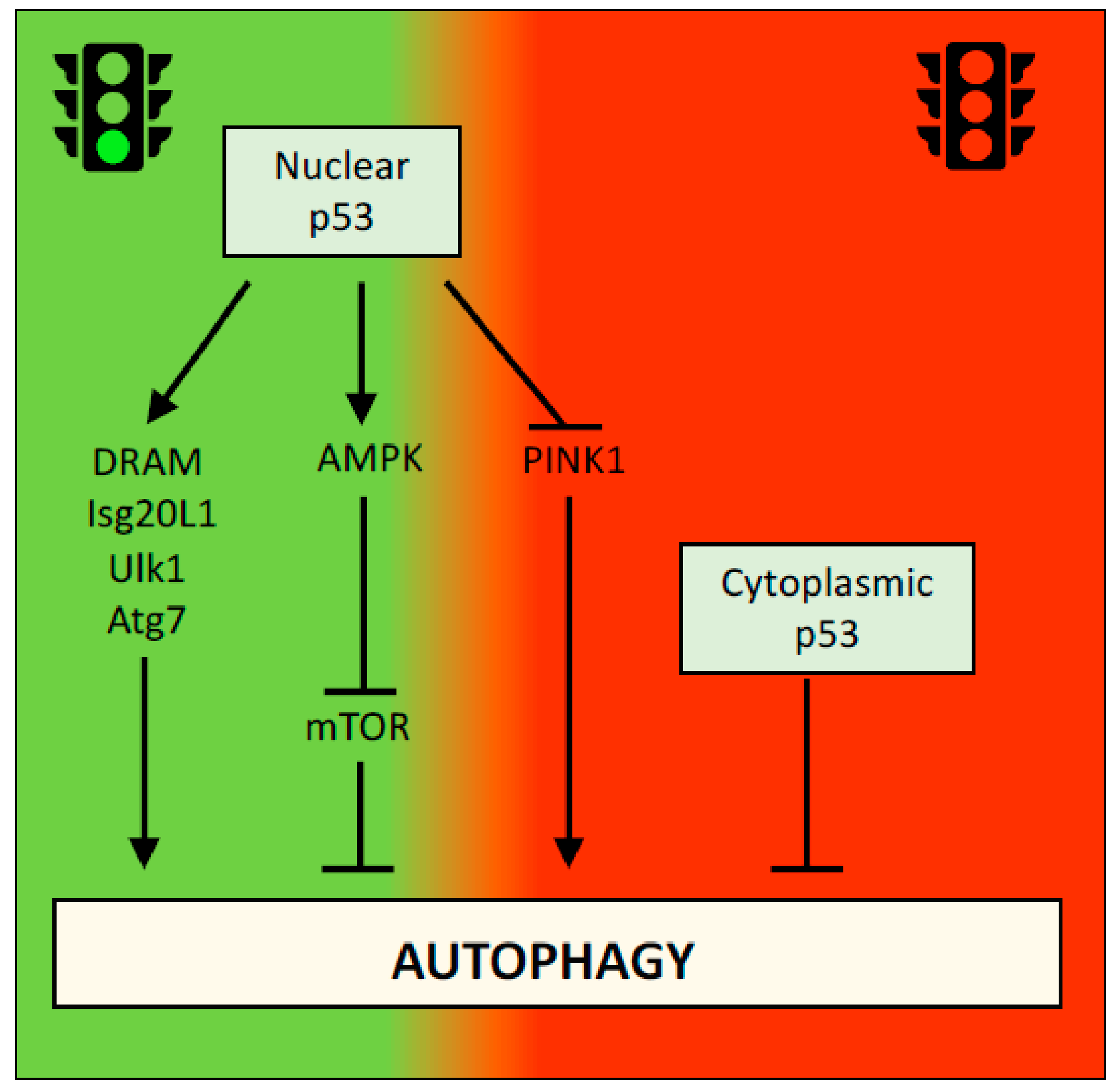
2.2. p53 and Autophagy
2.3. p53 Opposes EMT and Cell Migration
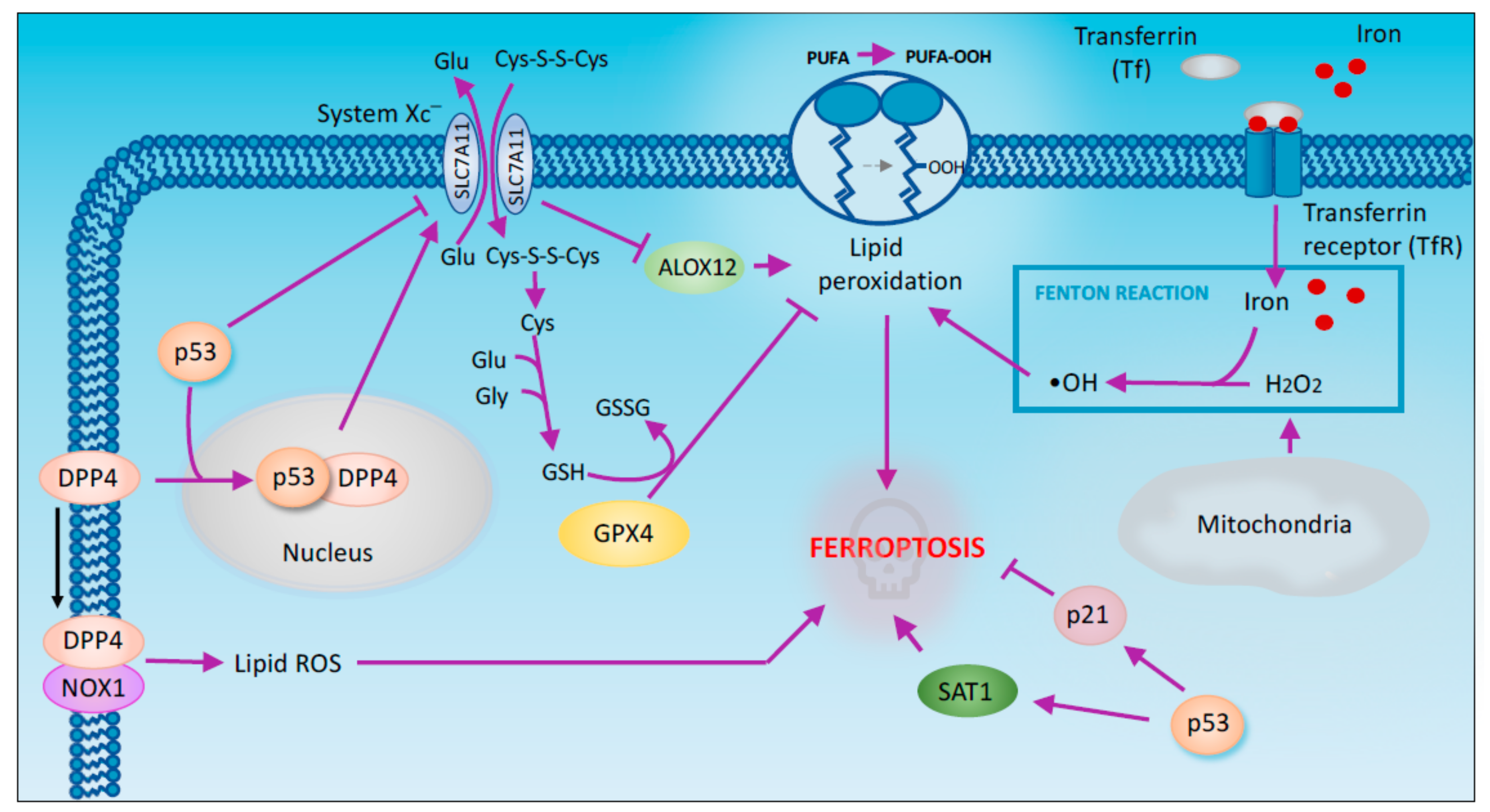
2.4. Ferroptosis
2.5. Maintenance of Genome Stability
2.6. Non-Coding RNA Regulated by p53 in Tumor Suppression
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Charni, M.; Aloni-Grinstein, R.; Molchadsky, A.; Rotter, V. p53 on the crossroad between regeneration and cancer. Cell Death Differ. 2017, 24, 8–14. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 2018, 20, 28–35. [Google Scholar] [CrossRef]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; Le Quesne, J.; Baena Acevedo, J.D.; et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. p53 ubiquitination: Mdm2 and beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef]
- Wu, D.; Prives, C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 2018, 25, 169–179. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and consequences. Cancers (Basel) 2014, 7, 30–69. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Tonnessen-Murray, C.A.; Lozano, G.; Jackson, J.G. The Regulation of cellular functions by the p53 Protein: Cellular senescence. Cold Spring Harb. Perspect. Med. 2017, 7, a026112. [Google Scholar] [CrossRef]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Kaiser, A.M.; Attardi, L.D. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018, 25, 93–103. [Google Scholar] [CrossRef]
- Clarke, A.R.; Purdie, C.A.; Harrison, D.J.; Morris, R.G.; Bird, C.C.; Hooper, M.L.; Wyllie, A.H. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 1993, 362, 849–852. [Google Scholar] [CrossRef]
- Lowe, S.W.; Schmitt, E.M.; Smith, S.W.; Osborne, B.A.; Jacks, T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993, 362, 847–849. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A two-faced genome guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Nemajerova, A.; Amelio, I.; Gebel, J.; Dotsch, V.; Melino, G.; Moll, U.M. Non-oncogenic roles of TAp73: From multiciliogenesis to metabolism. Cell Death Differ. 2018, 25, 144–153. [Google Scholar] [CrossRef]
- Sengupta, S.; Harris, C.C. p53: Traffic cop at the crossroads of DNA repair and recombination. Nat. Rev. Mol. Cell Biol 2005, 6, 44–55. [Google Scholar] [CrossRef]
- Smith, M.L.; Chen, I.T.; Zhan, Q.; Bae, I.; Chen, C.Y.; Gilmer, T.M.; Kastan, M.B.; O’Connor, P.M.; Fornace, A.J., Jr. Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 1994, 266, 1376–1380. [Google Scholar] [CrossRef]
- Sturzbecher, H.W.; Donzelmann, B.; Henning, W.; Knippschild, U.; Buchhop, S. p53 is linked directly to homologous recombination processes via RAD51/RecA protein interaction. EMBO J. 1996, 15, 1992–2002. [Google Scholar] [CrossRef]
- Linke, S.P.; Sengupta, S.; Khabie, N.; Jeffries, B.A.; Buchhop, S.; Miska, S.; Henning, W.; Pedeux, R.; Wang, X.W.; Hofseth, L.J.; et al. p53 interacts with hRAD51 and hRAD54, and directly modulates homologous recombination. Cancer Res. 2003, 63, 2596–2605. [Google Scholar]
- Gottifredi, V.; Wiesmuller, L. The tip of an iceberg: Replication-associated functions of the tumor suppressor p53. Cancers 2018, 10, 250. [Google Scholar] [CrossRef]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef]
- Mello, S.S.; Attardi, L.D. Deciphering p53 signaling in tumor suppression. Curr. Opin. Cell Biol. 2018, 51, 65–72. [Google Scholar] [CrossRef]
- Valente, L.J.; Gray, D.H.; Michalak, E.M.; Pinon-Hofbauer, J.; Egle, A.; Scott, C.L.; Janic, A.; Strasser, A. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013, 3, 1339–1345. [Google Scholar] [CrossRef]
- Teoh, S.T.; Lunt, S.Y. Metabolism in cancer metastasis: Bioenergetics, biosynthesis, and beyond. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1406. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. P53 regulates glucose metabolism through an IKK-NF-kappa B pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Ros, S.; Floter, J.; Kaymak, I.; Da Costa, C.; Houddane, A.; Dubuis, S.; Griffiths, B.; Mitter, R.; Walz, S.; Blake, S.; et al. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 is essential for p53-null cancer cells. Oncogene 2017, 36, 3287–3299. [Google Scholar] [CrossRef]
- Contractor, T.; Harris, C.R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of AIF expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef]
- Morris, J.P.t.; Yashinskie, J.J.; Koche, R.; Chandwani, R.; Tian, S.; Chen, C.C.; Baslan, T.; Marinkovic, Z.S.; Sanchez-Rivera, F.J.; Leach, S.D.; et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019, 573, 595–599. [Google Scholar] [CrossRef]
- Levine, A.J.; Berger, S.L. The interplay between epigenetic changes and the p53 protein in stem cells. Genes Dev. 2017, 31, 1195–1201. [Google Scholar] [CrossRef]
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mak, T.W.; Benchimol, S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 2011, 44, 491–501. [Google Scholar] [CrossRef]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Nakagawa, Y.; Ide, T.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. p53 Activation in adipocytes of obese mice. J. Biol. Chem. 2003, 278, 25395–25400. [Google Scholar] [CrossRef]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.T.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell 2019, 176, 564–580. [Google Scholar] [CrossRef]
- Li, L.; Mao, Y.; Zhao, L.; Li, L.; Wu, J.; Zhao, M.; Du, W.; Yu, L.; Jiang, P. p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 2019, 567, 253–256. [Google Scholar] [CrossRef]
- Feng, Y.; Yao, Z.; Klionsky, D.J. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015, 25, 354–363. [Google Scholar] [CrossRef]
- Klionsky, D.J. Why do we need autophagy? A cartoon depiction. Autophagy 2018, 14, 739–742. [Google Scholar] [CrossRef]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef]
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef]
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef]
- Eby, K.G.; Rosenbluth, J.M.; Mays, D.J.; Marshall, C.B.; Barton, C.E.; Sinha, S.; Johnson, K.N.; Tang, L.; Pietenpol, J.A. ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Mol. Cancer 2010, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef]
- Goiran, T.; Duplan, E.; Rouland, L.; El Manaa, W.; Lauritzen, I.; Dunys, J.; You, H.; Checler, F.; Alves da Costa, C. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death Differ. 2018, 25, 873–884. [Google Scholar] [CrossRef]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.S.; et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef]
- Termen, S.; Tan, E.J.; Heldin, C.H.; Moustakas, A. p53 regulates epithelial-mesenchymal transition induced by transforming growth factor beta. J. Cell Physiol. 2013, 228, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.O.; Kim, H.; Jung, G. p53 inhibits tumor cell invasion via the degradation of snail protein in hepatocellular carcinoma. FEBS Lett. 2010, 584, 2231–2236. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.K.; Eves, R.; Jia, L.; Mooney, P.; Mak, A.S. p53 suppresses Src-induced podosome and rosette formation and cellular invasiveness through the upregulation of caldesmon. Mol. Cell Biol. 2009, 29, 3088–3098. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, M.; Elia, L.; Condorelli, G.; Courtneidge, S.A. MicroRNA control of podosome formation in vascular smooth muscle cells in vivo and in vitro. J. Cell Biol. 2010, 189, 13–22. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef]
- Lefort, K.; Mandinova, A.; Ostano, P.; Kolev, V.; Calpini, V.; Kolfschoten, I.; Devgan, V.; Lieb, J.; Raffoul, W.; Hohl, D.; et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007, 21, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Furth, N.; Aylon, Y.; Oren, M. p53 shades of Hippo. Cell Death Differ. 2018, 25, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Valente, L.J.; Raj, N.; Seoane, J.A.; Flowers, B.M.; McClendon, J.; Bieging-Rolett, K.T.; Lee, J.; Ivanochko, D.; Kozak, M.M.; et al. A p53 super-tumor suppressor reveals a tumor suppressive p53-Ptpn14-Yap axis in pancreatic cancer. Cancer Cell 2017, 32, 460–473. [Google Scholar] [CrossRef]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Fearnhead, H.O.; Vandenabeele, P.; Vanden Berghe, T. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ. 2017, 24, 1991–1998. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579. [Google Scholar] [CrossRef]
- Casero, R.A.; Stewart, T.M.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.J.; Li, D.W.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z.H. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Amelio, I.; Markert, E.K.; Rufini, A.; Antonov, A.V.; Sayan, B.S.; Tucci, P.; Agostini, M.; Mineo, T.C.; Levine, A.J.; Melino, G. p73 regulates serine biosynthesis in cancer. Oncogene 2014, 33, 5039–5046. [Google Scholar] [CrossRef]
- Giacobbe, A.; Bongiorno-Borbone, L.; Bernassola, F.; Terrinoni, A.; Markert, E.K.; Levine, A.J.; Feng, Z.H.; Agostini, M.; Zolla, L.; Agro, A.F.; et al. p63 regulates glutaminase 2 expression. Cell Cycle 2013, 12, 1395–1405. [Google Scholar] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Gao, M.H.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X.J. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Hayashi, M.; Madokoro, H.; Nishida, H.; Du, W.L.; Ohnuma, K.; Sakamoto, M.; Morimoto, C.; Yamada, T. Nuclear localization of CD26 induced by a humanized monoclonal antibody inhibits tumor cell growth by modulating of POLR2A transcription. PLoS ONE 2013, 8, e62304. [Google Scholar] [CrossRef]
- Xie, Y.C.; Zhu, S.; Song, X.X.; Sun, X.F.; Fan, Y.; Liu, J.B.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.B.; Attardi, L.D.; Dixon, S.J. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef]
- Toufektchan, E.; Toledo, F. The guardian of the genome revisited: p53 downregulates genes required for telomere maintenance, DNA repair, and centromere structure. Cancers 2018, 10, 135. [Google Scholar] [CrossRef]
- Liu, G.; Parant, J.M.; Lang, G.; Chau, P.; Chavez-Reyes, A.; El-Naggar, A.K.; Multani, A.; Chang, S.; Lozano, G. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat. Genet. 2004, 36, 63–68. [Google Scholar] [CrossRef]
- Levine, A.J.; Ting, D.T.; Greenbaum, B.D. P53 and the defenses against genome instability caused by transposons and repetitive elements. Bioessays 2016, 38, 508–513. [Google Scholar] [CrossRef]
- Wylie, A.; Jones, A.E.; D’Brot, A.; Lu, W.J.; Kurtz, P.; Moran, J.V.; Rakheja, D.; Chen, K.S.; Hammer, R.E.; Comerford, S.A.; et al. p53 genes function to restrain mobile elements. Genes Dev. 2016, 30, 64–77. [Google Scholar] [CrossRef]
- Wylie, A.; Jones, A.E.; Abrams, J.M. p53 in the game of transposons. Bioessays 2016, 38, 1111–1116. [Google Scholar] [CrossRef]
- Dudgeon, C.; Chan, C.; Kang, W.F.; Sun, Y.; Emerson, R.; Robins, H.; Levine, A.J. The evolution of thymic lymphomas in p53 knockout mice. Gene Dev. 2014, 28, 2613–2620. [Google Scholar] [CrossRef]
- He, L.; He, X.Y.; Lim, L.P.; De Stanchina, E.; Xuan, Z.Y.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130. [Google Scholar] [CrossRef]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef]
- Handa, H.; Murakami, Y.; Ishihara, R.; Kimura-Masuda, K.; Masuda, Y. The role and function of microRNA in the pathogenesis of multiple myeloma. Cancers 2019, 11, 1738. [Google Scholar] [CrossRef]
- Chiang, Y.P.; Song, Y.X.; Wang, Z.N.; Liu, Z.K.; Gao, P.; Liang, J.W.; Zhu, J.L.; Xing, C.Z.; Xu, H.M. microRNA-192,-194 and-215 are frequently downregulated in colorectal cancer. Exp. Ther Med. 2012, 3, 560–566. [Google Scholar] [CrossRef]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Lotterman, C.D.; Bao, C.; Hruban, R.; Karim, B.; Mendell, J.T.; Huso, D.; Lowenstein, C.J. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6334–6339. [Google Scholar] [CrossRef]
- Liang, M.; Yao, G.D.; Yin, M.M.; Lu, M.R.; Tian, H.; Liu, L.; Lian, J.; Huang, X.X.; Sun, F. Transcriptional cooperation between p53 and NF-kappa B p65 regulates microRNA-224 transcription in mouse ovarian granulosa cells. Mol. Cell Endocrinol. 2013, 370, 119–129. [Google Scholar] [CrossRef]
- Yan, H.L.; Xue, G.; Mei, Q.; Wang, Y.Z.; Ding, F.X.; Liu, M.F.; Lu, M.H.; Tang, Y.; Yu, H.Y.; Sun, S.H. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. Embo J. 2009, 28, 2719–2732. [Google Scholar] [CrossRef]
- Hu, W.L.; Jin, L.; Xu, A.; Wang, Y.F.; Thorne, R.F.; Zhang, X.D.; Wu, M.A. GUARDIN is a p53-responsive long non-coding RNA that is essential for genomic stability. Nat. Cell Biol. 2018, 20, 492. [Google Scholar] [CrossRef]
- Mello, S.S.; Sinow, C.; Raj, N.; Mazur, P.K.; Bieging-Rolett, K.; Broz, D.K.; Imam, J.F.C.; Vogel, H.; Wood, L.D.; Sage, J.; et al. Neat1 is a p53-inducible lincRNA essential for transformation suppression. Gene Dev. 2017, 31, 1095–1108. [Google Scholar] [CrossRef]
- Puvvula, P.K.; Desetty, R.D.; Pineau, P.; Marchio, A.; Moon, A.; Dejean, A.; Bischof, O. Long noncoding RNA PANDA and scaffold-attachment-factor SAFA control senescence entry and exit. Nat. Commun 2014, 5, 5253. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p53 Upregulated miRs | Pathways | Targets |
| miR-34a/b/c | cell proliferation, EMT | cyclin E2, CDK4, CDK6, BCL2, SNAIL1 |
| miR-145 | cell growth | c-Myc |
| miR-192 | EMT | ZEB2 |
| miR-194/miR-215 | DNA synthesis, cell cycle | CUL5, LMNB2, CDC7, MAD2L1, BCL2, |
| miR-200 | EMT | ZEB1/2 |
| miR-107 | hypoxia response | HIF1β |
| p53 Downregulated miRs | Pathways | Targets |
| miR-224 | cell proliferation | SMAD4 |
| miR-17-92 | apoptosis, cell proliferation | Bim, EGR2, p21 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitolli, C.; Wang, Y.; Candi, E.; Shi, Y.; Melino, G.; Amelio, I. p53-Mediated Tumor Suppression: DNA-Damage Response and Alternative Mechanisms. Cancers 2019, 11, 1983. https://doi.org/10.3390/cancers11121983
Pitolli C, Wang Y, Candi E, Shi Y, Melino G, Amelio I. p53-Mediated Tumor Suppression: DNA-Damage Response and Alternative Mechanisms. Cancers. 2019; 11(12):1983. https://doi.org/10.3390/cancers11121983
Chicago/Turabian StylePitolli, Consuelo, Ying Wang, Eleonora Candi, Yufang Shi, Gerry Melino, and Ivano Amelio. 2019. "p53-Mediated Tumor Suppression: DNA-Damage Response and Alternative Mechanisms" Cancers 11, no. 12: 1983. https://doi.org/10.3390/cancers11121983
APA StylePitolli, C., Wang, Y., Candi, E., Shi, Y., Melino, G., & Amelio, I. (2019). p53-Mediated Tumor Suppression: DNA-Damage Response and Alternative Mechanisms. Cancers, 11(12), 1983. https://doi.org/10.3390/cancers11121983