Promising Colorectal Cancer Biomarkers for Precision Prevention and Therapy

 ,
, 
Abstract
1. Introduction
2. Colorectal Cancer: An Overview
3. Role of Molecular Biomarkers in CRC Management
4. Molecular Features of Hereditary Colorectal Cancer
5. Role of Molecular Biomarkers in the Surgical Approach to Hereditary Colorectal Cancers
6. Predictive Biomarkers in CRC Therapy and Prognosis
7. Future Perspectives in the Field of Cancer Biomarkers
7.1. Molecular Subtypes
7.2. Circulating Biomarkers
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.; Rega, D.; Costabile, V.; Duraturo, F.; Niglio, A.; Izzo, P.; Pace, U.; Delrio, P. The biological complexity of colorectal cancer: Insights into biomarkers for early detection and personalized care. Ther. Adv. Gastroenterol. 2016, 9, 861–886. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.; Pace, U.; Rega, D.; Costabile, V.; Duraturo, F.; Izzo, P.; Delrio, P. Genetics, diagnosis and management of colorectal cancer. Oncol. Rep. 2015, 34, 1087–1096. [Google Scholar] [CrossRef]
- Armelao, F.; de Pretis, G. Familial colorectal cancer: A review. World J. Gastroenterol. 2014, 20, 9292–9298. [Google Scholar] [CrossRef]
- Fanali, C.; Lucchetti, D.; Farina, M.; Corbi, M.; Cufino, V.; Cittadini, A.; Sgambato, A. Cancer stem cells in colorectal cancer from pathogenesis to therapy: Controversies and perspectives. World J. Gastroenterol. 2014, 20, 923–942. [Google Scholar] [CrossRef]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2017, 9, 33403–33415. [Google Scholar] [CrossRef]
- Ong, B.A.; Vega, K.J.; Houchen, C.W. Intestinal stem cells and the colorectal cancer microenvironment. World J. Gastroenterol. 2014, 20, 1898–1909. [Google Scholar]
- Sideris, M.; Papagrigoriadis, S. Molecular biomarkers and classification models in the evaluation of the prognosis of colorectal cancer. Anticancer Res. 2014, 34, 2061–2068. [Google Scholar]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Colussi, D.; Brandi, G.; Bazzoli, F.; Ricciardiello, L. Molecular pathways involved in colorectal cancer: Implications for disease behavior and prevention. Int. J. Mol. Sci. 2013, 14, 16365–16385. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, L.A.; Peltomäki, P.; Leach, F.S.; Sistonen, P.; Pylkkänen, L.; Mecklin, J.P.; Järvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, S.N.; French, A.J.; Roche, P.C.; Cunningham, J.M.; Tester, D.J.; Lindor, N.M.; Moslein, G.; Baker, S.M.; Liskay, R.M.; Burgart, L.J.; et al. Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res. 1996, 56, 4836–4840. [Google Scholar]
- Vasen, H.F.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007, 50, 113–130. [Google Scholar] [CrossRef]
- Duraturo, F.; Liccardo, R.; Cavallo, A.; De Rosa, M.; Rossi, G.B.; Izzo, P. Multivariate analysis as a method for evaluating the pathogenicity of novel genetic MLH1 variants in patients with colorectal cancer and microsatellite instability. Int. J. Mol. Med. 2015, 36, 511–517. [Google Scholar] [CrossRef]
- Carethers, J.M.; Koi, M.; Tseng-Rogenski, S.S. EMAST is a form of microsatellite instability that is initiated by inflammation and modulates colorectal cancer progression. Genes 2015, 6, 185–205. [Google Scholar] [CrossRef]
- Huang, S.C.; Lee, J.K.; Smith, E.J.; Doctolero, R.T.; Tajima, A.; Beck, S.E.; Weidner, N.; Carethers, J.M. Evidence for an hMSH3 defect in familial hamartomatous polyps. Cancer 2011, 117, 492–500. [Google Scholar] [CrossRef]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Choi, C.; Kim, H.R.; Daoud, Y.; Toiyama, Y.; Takahashi, M.; Goel, A.; Boland, C.R.; Koi, M. Association between recurrent metastasis from stage II and III primary colorectal tumors and moderate microsatellite instability. Gastroenterology 2012, 143, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Tseng-Rogenski, S.S.; Chung, H.; Wilk, M.B.; Zhang, S.; Iwaizumi, M.; Carethers, J.M. Oxidative stress induces nuclear-to-cytosol shift of hMSH3, a potential mechanism for EMAST in colorectal cancer cells. PLoS ONE 2012, 7, e50616. [Google Scholar] [CrossRef] [PubMed]
- Campregher, C.; Schmid, G.; Ferk, F.; Knasmüller, S.; Khare, V.; Kortüm, B.; Dammann, K.; Lang, M.; Scharl, T.; Spittler, A.; et al. MSH3-deficiency initiates EMAST without oncogenic transformation of human colon epithelial cells. PLoS ONE 2012, 7, e50541. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef]
- Duval, A.; Hamelin, R. Genetic instability in human mismatch repair deficient cancers. Ann. Genet. 2002, 45, 71–75. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Cuilliere-Dartigues, P.; El-Bchiri, J.; Krimi, A.; Buhard, O.; Fontanges, P.; Fléjou, J.F.; Hamelin, R.; Duval, A. TCF-4 isoforms absent in TCF-4 mutated MSI-H colorectal cancer cells colocalize with nuclear CtBP and repress TCF-4-mediated transcription. Oncogene 2006, 25, 4441–4448. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Fang, M.; Hutchinson, L.; Deng, A.; Green, M.R. Common BRAF(V600E)-directed pathway mediates widespread epigenetic silencing in colorectal cancer and melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, 1250–1255. [Google Scholar] [CrossRef]
- Capper, D.; Voigt, A.; Bozukova, G.; Ahadova, A.; Kickingereder, P.; von Deimling, A.; von Knebel Doeberitz, M.; Kloor, M. BRAF V600E-specific immunohistochemistry for the exclusion of Lynch syndrome in MSI-H colorectal cancer. Int. J. Cancer 2013, 133, 1624–1630. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.A.; Wu, C.; Yu, M.; Gourgioti, G.; Wirtz, R.; Raptou, G.; Gkakou, C.; Kotoula, V.; Pentheroudakis, G.; Papaxoinis, G. Evaluation of CpG Island Methylator Phenotype as a Biomarker in Colorectal Cancer Treated With Adjuvant Oxaliplatin. Clin. Colorectal Cancer 2016, 15, 164–169. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vermeulen, L.; De Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Espejo-Herrera, N.; Gràcia-Lavedan, E.; Boldo, E.; Aragonés, N.; Pérez-Gómez, B.; Pollán, M.; Molina, A.J.; Fernández, T.; Martín, V.; La Vecchia, C.; et al. Colorectal cancer risk and nitrate exposure through drinking water and diet. Int. J. Cancer 2016, 139, 334–346. [Google Scholar] [CrossRef]
- Tang, X. Tumor-associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. 2013, 332, 3–10. [Google Scholar] [CrossRef]
- Xuan, Q.J.; Wang, J.X.; Nanding, A.; Wang, Z.P.; Liu, H.; Lian, X.; Zhang, Q.Y. Tumor-associated macrophages are correlated with tamoxifen resistance in the postmenopausal breast cancer patients. Pathol. Oncol. Res. 2014, 20, 619–624. [Google Scholar] [CrossRef]
- Yuxin, L.; Jianxin, X.; Huiyin, L. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar]
- Yoon, N.; Han, K.M.; Cho, S.Y.; Kim, S.W.; Lee, J.E.; Nam, S.J.; Cho, E.Y. Tumor-associated macrophages (TAMs) and tumor-infiltrating lymphocytes (TILs) in pretherapeutic breast cancer core biopsies: Anti-tumoral effect of immune cells associated with neoadjuvant chemotherapy. Int. J. Clin. Exp. Pathol. 2017, 10, 1738–1746. [Google Scholar]
- Clarke, W.T.; Feuerstein, J.D. Colorectal cancer surveillance in inflammatory bowel disease: Practice guidelines and recent developments. World J. Gastroenterol. 2019, 25, 4148–4157. [Google Scholar] [CrossRef]
- Sing Vink, G.; Jafri, M.; Mehdi, S.; Ashley, C. Staging and survival of colorectal cancer (CRC) in octogenarians: Nationwide Study of US Veterans. J. Gastrointest. Oncol. 2019, 10, 12–18. [Google Scholar] [CrossRef]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Tveit, K.M.; Guren, T.; Glimelius, B.; Pfeiffer, P.; Sorbye, H.; Pyrhonen, S.; Sigurdsson, F.; Kure, E.; Ikdahl, T.; Skovlund, E. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: The NORDIC-VII study. J. Clin. Oncol. 2012, 30, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Price, T.J.; Cervantes, A.; Sobrero, A.F.; Ducreux, M.; Hotko, Y.; André, T.; Chan, E.; Lordick, F.; Punt, C.J.; et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 4706–4713. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Duraturo, F.; Liccardo, R.; De Rosa, M.; Izzo, P. Genetics, diagnosis and treatment of Lynch syndrome: Old lessons and current challenges. Oncol. Lett. 2019, 17, 3048–3054. [Google Scholar] [CrossRef]
- Sargent, D.; Sobrero, A.; Grothey, A.; O’Connell, M.J.; Buyse, M.; Andre, T.; Zheng, Y.; Green, E.; Labianca, R.; O’Callaghan, C.; et al. Evidence for cure by adjuvant therapy in colon cancer: Observations based on individual patient data from 20,898 patients on 18 randomized trials. J. Clin. Oncol. 2009, 27, 872–877. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Siegel, R.; Desantis, C.; Jemal, A. Colorectal cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 104–117. [Google Scholar] [CrossRef]
- Edwards, B.K.; Ward, E.; Kohler, B.A.; Eheman, C.; Zauber, A.G.; Anderson, R.N.; Jemal, A.; Schymura, M.J.; Lansdorp-Vogelaar, I.; Seeff, L.C.; et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010, 116, 544–573. [Google Scholar] [CrossRef]
- Adler, A.; Geiger, S.; Keil, A.; Bias, H.; Schatz, P.; deVos, T.; Dhein, J.; Zimmermann, M.; Tauber, R.; Wiedenmann, B. Improving compliance to colorectal cancer screening using blood and stool based tests in patients refusing screening colonoscopy in Germany. BMC Gastroenterol. 2014, 14, 183. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; Vilar, E.; Tavtigianand, S.V.; Stoffel, E.M. Genetic predisposition to colorectal cancer: Syndromes, genes, classification of genetic variants and implications for precision medicine. J. Pathol. 2019, 247, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.M.; Koeppe, E.; Everett, J.; Ulintz, P.; Kiel, M.; Osborne, J.; Williams, L.; Hanson, K.; Gruber, S.B.; Rozek, L.S. Germline Genetic Features of Young Individuals With Colorectal Cancer. Gastroenterology 2018, 154, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Mork, M.E.; You, Y.N.; Ying, J.; Bannon, S.A.; Lynch, P.M.; Rodriguez-Bigas, M.A.; Vilar, E. High Prevalence of Hereditary Cancer Syndromes in Adolescents and Young Adults With Colorectal Cancer. J. Clin. Oncol. 2015, 33, 3544–3549. [Google Scholar] [CrossRef]
- Samadder, N.J.; Kuwada, S.K.; Boucher, K.M.; Byrne, K.; Kanth, P.; Samowitz, W.; Jones, D.; Tavtigian, S.V.; Westover, M.; Berry, T.; et al. Association of Sulindac and Erlotinib vs Placebo With Colorectal Neoplasia in Familial Adenomatous Polyposis Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2018, 4, 671–677. [Google Scholar] [CrossRef]
- Barnard, J. Screening and Surveillance Recommendations for Pediatric Gastrointestinal Polyposis Syndromes. J. Pediatr. Gastroenterol. Nutr. 2009, 48 (Suppl. 2), S75–S78. [Google Scholar] [CrossRef]
- Talseth-Palmer, B. The genetic basis of colonic adenomatous polyposis syndromes. Hered. Cancer Clin. Pract. 2017, 15, 5. [Google Scholar] [CrossRef]
- Esteban-Jurado, C.; Giménez-Zaragoza, D.; Muñoz, J.; Franch-Expósito, S.; Álvarez-Barona, M.; Ocaña, T.; Cuatrecasas, M.; Carballal, S.; López-Cerón, M.; Marti-Solano, M. POLE and POLD1 screening in 155 patients with multiple polyps and early-onset colorectal cancer. Oncotarget 2017, 8, 26732–26743. [Google Scholar] [CrossRef]
- Lubbe, S.J.; Di Bernardo, M.C.; Chandler, I.P.; Houlston, R.S. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J. Clin. Oncol. 2009, 27, 3975–3980. [Google Scholar] [CrossRef]
- Boland, P.M.; Yurgelun, M.B.; Boland, R.C. Recent Progress in Lynch Syndrome and Other Familial Colorectal Cancer Syndromes. CA Cancer J. Clin. 2018, 68, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. Microsatellite Instability Pathway and EMAST in Colorectal Cancer. Curr. Colorectal Cancer Rep. 2017, 13, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Brosens, L.A.; van Hattem, A.; Hylind, L.M.; Iacobuzio-Donahue, C.; Romans, K.E.; Axilbund, J.; Cruz-Correa, M.; Tersmette, A.C.; Offerhaus, G.J.; Giardiello, F.M. Risk of colorectal cancer in juvenile polyposis. Gut 2007, 56, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.N.; Kopetz, E.S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: Clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 2015, 6, 660–667. [Google Scholar] [CrossRef]
- Rosty, C.; Buchanan, D.D.; Walsh, M.D.; Pearson, S.A.; Pavluk, E.; Walters, R.J.; Clendenning, M.; Spring, K.J.; Jenkins, M.A.; Win, A.K. Phenotype and polyp landscape in serrated polyposis syndrome: A series of 100 patients from genetics clinics. Am. J. Surg. Pathol. 2012, 36, 876. [Google Scholar] [CrossRef]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes. Am. J. Gastroenterol. 2015, 110, 223–263. [Google Scholar] [CrossRef]
- Nieminen, T.T.; O’Donohue, M.F.; Wu, Y.; Lohi, H.; Scherer, S.W.; Paterson, A.D.; Ellonen, P.; Abdel-Rahman, W.M.; Valo, S.; Mecklin, J.P.; et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology 2014, 147, 595–598. [Google Scholar] [CrossRef]
- Herzig, D.O.; Buie, W.D.; Weiser, M.R.; You, Y.N.; Rafferty, J.F.; Feingold, D.; Steele, S.R. Clinical Practice Guidelines for the Surgical Treatment of Patients With Lynch Syndrome. Dis. Colon Rectum 2017, 60, 137–143. [Google Scholar] [CrossRef]
- Sepulveda, A.R.; Hamilton, S.R.; Allegra, C.J.; Grody, W.; Cushman-Vokoun, A.M.; Funkhouser, W.K.; Kopetz, S.E.; Lieu, C.; Lindor, N.M.; Minsky, B.D.; et al. Molecular Biomarkers for the Evaluation of Colorectal Cancer: Guideline From the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and American Society of Clinical Oncology. J. Mol. Diagn. 2017, 19, 187–225. [Google Scholar] [CrossRef]
- Herzig, D.; Hardiman, K.; Weiser, M.; You, N.; Paquette, I.; Feingold, D.L.; Steele, S.R. The American Society of Colon and Rectal Surgeons Clinical Practice Guidelines for the Management of Inherited Polyposis Syndromes. Dis. Colon Rectum 2017, 60, 881–894. [Google Scholar] [CrossRef]
- Nieuwenhuis, M.H.; Douma, K.F.; Bleiker, E.M.; Bemelman, W.A.; Aaronson, N.K.; Vasen, H.F. Female fertility after colorectal surgery for familial adenomatous polyposis: A nationwide cross-sectional study. Ann. Surg. 2010, 252, 341–344. [Google Scholar] [CrossRef]
- Dodaro, C.; Grifasi, C.; Florio, J.; Santangelo, M.L.; Duraturo, F.; De Rosa, M.; Izzo, P.; Renda, A. The role of mutation analysis of the APC gene in the management of FAP patients. A controversial issue. Ann. Ital. Chir. 2016, 87, 321–325. [Google Scholar] [PubMed]
- Dicks, E.; Pullman, D.; Kao, K.; MacMillan, A.; Simmonds, C.; Etchegary, H. Universal tumor screening for Lynch syndrome: Perspectives of Canadian pathologists and genetic counselors. Commun. Genet. 2019, 10, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.X.; Tang, Q.Y.; Bai, J.L.; Qian, X.P.; Li, R.T.; Liu, B.R.; Zheng, M.H. Predictive value of thymidylate synthase expression in advanced colorectal cancer patients receiving fluoropyrimidine-based chemotherapy: Evidence from 24 studies. Int. J. Cancer 2008, 123, 2384–2389. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, E.A.; Fanelli, M.F.; Buim, M.E.; Machado Netto, M.C.; Gasparini Junior, J.L.; Souza ESilva, V.; Dettino, A.L.; Mingues, N.B.; Romero, J.V.; Ocea, L.M.; et al. Thymidylate synthase expression in circulating tumor cells: A new tool to predict 5-fluorouracil resistance in metastatic colorectal cancer patients. Int. J. Cancer 2015, 137, 1397–1405. [Google Scholar] [CrossRef]
- Zhou, J.Y.; Shi, R.; Yu, H.L.; Zeng, Y.; Zheng, W.L.; Ma, W.L. The association between two polymorphisms in the TS gene and risk of cancer: A systematic review and pooled analysis. Int. J. Cancer 2012, 131, 2103–2116. [Google Scholar] [CrossRef]
- Stark, M.; Bram, E.E.; Akerman, M.; Mandel-Gutfreund, Y.; Assaraf, Y.G. Heterogeneous nuclear ribonucleoprotein H1/H2-dependent unsplicing of thymidine phosphorylase results in anticancer drug resistance. J. Biol. Chem. 2011, 286, 3741–3754. [Google Scholar] [CrossRef]
- Lin, S.; Lai, H.; Qin, Y.; Chen, J.; Lin, Y. Thymidine phosphorylase and hypoxia-inducible factor 1-α expression in clinical stage II/III rectal cancer: Association with response to neoadjuvant chemoradiation therapy and prognosis. Int. J. Clin. Exp. Pathol. 2015, 8, 10680–10688. [Google Scholar]
- Gnoni, A.; Russo, A.; Silvestris, N.; Maiello, E.; Vacca, A.; Marech, I.; Numico, G.; Paradiso, A.; Lorusso, V.; Azzariti, A. Pharmacokinetic and metabolism determinants of fluoropyrimidines and oxaliplatin activity in treatment of colorectal patients. Curr. Drug Metab. 2011, 12, 918–931. [Google Scholar] [CrossRef]
- Zeuner, A.; Todaro, M.; Stassi, G.; De Maria, R. Colorectal cancer stem cells: From the crypt to the clinic. Cell Stem Cell 2014, 15, 692–705. [Google Scholar] [CrossRef]
- Turano, M.; Costabile, V.; Cerasuolo, A.; Duraturo, F.; Liccardo, R.; Delrio, P.; Pace, U.; Rega, D.; Dodaro, C.A.; Milone, M.; et al. Characterisation of mesenchymal colon tumour-derived cells in tumourspheres as a model for colorectal cancer progression. Int. J. Oncol. 2018, 53, 2379–2396. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.H.; Láng, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. 2011, 29, 2011–2019. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Jonker, D.J.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Osumi, H.; Shinozaki, E.; Osako, M.; Kawazoe, Y.; Oba, M.; Misaka, T.; Goto, T.; Kamo, H.; Suenaga, M.; Kumekawa, Y.; et al. Cetuximab treatment for metastatic colorectal cancer with KRAS p.G13D mutations improves progression-free survival. Mol. Clin. Oncol. 2015, 3, 1053–1057. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tejpar, S.; Celik, I.; Schlichting, M.; Sartorius, U.; Bokemeyer, C.; van Cutsem, E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J. Clin. Oncol. 2012, 30, 3570–3577. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.F.; McDermott, D.F.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1558–1568. [Google Scholar] [CrossRef]
- Rowland, A.; Dias, M.M.; Wiese, M.D.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Meta-analysis comparing the efficacy of anti-EGFR monoclonal antibody therapy between KRAS G13D and other KRAS mutant metastatic colorectal cancer tumours. Eur. J. Cancer 2016, 55, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, L.; Zhou, Q. Overall and KRAS- specific results of combined cetuximab treatment and chemotherapy for metastatic colorectal cancer: A meta-analysis. Int. J. Colorectal Dis. 2011, 26, 1025–1033. [Google Scholar] [CrossRef]
- Yin, W.H.; Fan, H.Z.; Sheng, J.W.; Xia, H.M.; Wu, Y.W.; Xie, P. Effect of vascular endothelial growth factor C and collagen triple helix repeat containing 1 expression on prognosis of rectal carcinoma patients. Chin. J. Gastrointest. Surg. 2013, 16, 673–675. [Google Scholar]
- Ciardiello, D.; Vitiello, P.P.; Cardone, C.; Martini, G.; Troiani, T.; Martinelli, E.; Ciardiello, F. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy T. Cancer Treat. Rev. 2019, 76, 22–23. [Google Scholar] [CrossRef]
- Cavnar, M.J.; Turcotte, S.; Katz, S.C.; Kuk, D.; Goönen, M.; Shia, J.; Allen, P.J.; Balachandran, V.P.; D’Angelica, M.I.; Kingham, T.P.; et al. Tumor-Associated Macrophage Infiltration in Colorectal Cancer Liver Metastases is Associated With Better Outcome. Ann. Surg. Oncol. 2017, 24, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Koelzer, V.H.; Canonica, K.; Dawson, H.; Sokol, L.; Karamitopoulou-Diamantis, E.; Lugli, A.; Zlobec, I. Phenotyping of tumor- associated macrophages in colorectal cancer: Impact on single cell invasion (tumor budding) and clinicopathological outcome. Oncoimmunol 2015, 5, e1106677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed]
- Malesci, A.; Bianchi, P.; Celesti, G.; Basso, G.; Marchesi, F.; Grizzi, F.; Di Caro, G.; Cavalleri, T.; Rimassa, L.; Palmqvist, R.; et al. Tumor- associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunol 2017, 6, e1342918. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Hurwitz, H. Combinations of bevacizumab with cancer immunotherapy. Cancer J. 2018, 24, 193–204. [Google Scholar] [CrossRef]
- Hegde, P.S.; Wallin, J.J.; Mancao, C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin. Cancer Biol. 2018, 52, 117–124. [Google Scholar] [CrossRef]
- Villadangos, J.A.; Schnorrer, P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol. 2007, 7, 543–555. [Google Scholar] [CrossRef]
- Oyama, T.; Ran, S.; Ishida, T.; Nadaf, S.; Kerr, L.; Carbone, D.P.; Gabrilovich, D.I. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-κB activation in hemopoietic progenitor cells. J. Immunol. 1998, 160, 1224. [Google Scholar]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in anti-angiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Ohm, J.E. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003, 101, 487886. [Google Scholar] [CrossRef] [PubMed]
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001, 91, 2417–2422. [Google Scholar] [CrossRef]
- Michael-Robinson, J.M.; Biemer-Hüttmann, A.; Purdie, D.M.; Walsh, M.D.; Simms, L.A.; Biden, K.G.; Young, J.P.; Leggett, B.A.; Jass, J.R.; Radford-Smith, G.L. Tumour infiltrating lymphocytes and apoptosis are independent features in colorectal cancer stratified according to microsatellite instability status. Gut 2001, 48, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, A.C.; Sørensen, F.B.; Lindebjerg, J.; Hager, H.; Christensen, R.; Frifeldt, S.K.; Hansen, T.F. The Prognostic Value of Tumor-Infiltrating lymphocytes in Stage II Colon Cancer. A Nationwide Population-Based Study. Transl. Oncol. 2018, 11, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.; Vreeland, T.; Trappey, A.; Hale, D.; Peace, K.; Tyler, J.; Walker, A.; Brown, R.; Herbert, G.; Yi, F.; et al. Cancer vaccines in colon and rectal cancer over the last decade: Lessons learned and future directions. Exp. Rev. Clin. Immunol. 2017, 13, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Caballero-Banños, M.; Benitez-Ribas, D.; Tabera, J.; Varea, S.; Vilana, R.; Bianchi, L.; Ayuso, J.R.; Pagés, M.; Carrera, G.; Cuatrecasas, M.; et al. Phase II randomised trial of autologous tumour lysate dendritic cell plus best supportive care compared with best supportive care in pre-treated advanced col- orectal cancer patients. Eur. J. Cancer 2016, 64, 167–174. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Claessen, A.M.; van Tinteren, H.; Gall, H.E.; Ezinga, R.; Meijer, S.; Scheper, R.J.; Meijer, C.J.L.M.; Bloemena, E.; Ransom, J.H.; et al. Active specific immunotherapy for stage II and stage III human colon cancer: A randomised trial. Lancet 1999, 353, 345–350. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Neal, R.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M.; et al. Line nivolumab plus ipilimumab in advanced non–small-cell lung cancer (CheckMate 568): Outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. JCO 2019, 37, 992–1000. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune land- scape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pageès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960. [Google Scholar] [CrossRef]
- Watanabe, T.; Wu, T.T.; Catalano, P.J.; Ueki, T.; Satriano, R.; Haller, D.G.; Benson, A.B.; Hamilton, S.R. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2001, 344, 1196–1206. [Google Scholar] [CrossRef]
- Lee, L.H.; Cavalcanti, M.S.; Segal, N.H.; Hechtman, J.F.; Weiser, M.R.; Smith, J.J.; Garcia, A.J.; Sadot, E.; Ntiamoah, P.; Markowitz, A.J.; et al. Patterns and prognostic relevance of PD-1 and PD-L1 expression in colorectal carcinoma. Mod. Pathol. 2016, 29, 1433–1442. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Okita, A.; Takahashi, S.; Ouchi, K.; Inoue, M.; Watanabe, M.; Endo, M.; Honda, H.; Yamada, Y.; Ishioka, C. Consensus molecular subtypes classification of colorectal cancer as a predictive factor for chemotherapeutic efficacy against metastatic colorectal cancer. Oncotarget 2018, 9, 18698–18711. [Google Scholar] [CrossRef]
- Johnson, K.R.; Wang, L.; Miller, M.C.; Willingham, M.C.; Fan, W. Fluorouracil interferes with paclitaxel cytotoxicity against human solid tumor cells. Clin. Cancer Res. 1997, 3, 1739–1745. [Google Scholar]
- Song, N.; Pogue-Geile, K.L.; Gavin, P.G.; Yothers, G.; Kim, S.R.; Johnson, N.L.; Lipchik, C.; Allegra, C.J.; Petrelli, N.J.; O’Connell, M.J.; et al. Clinical outcome from oxaliplatin treatment in stage II/III colon cancer according to intrinsic subtypes: Secondary analysis of NSABP C-07/NRG oncology randomized clinical trial. JAMA Oncol. 2016, 2, 1162–1169. [Google Scholar] [CrossRef]
- Isella, C.; Terrasi, A.; Bellomo, S.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.L.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Zanella, E.R.; Galimi, F.; Sassi, F.; Migliardi, G.; Cottino, F.; Leto, S.M.; Lupo, B.; Erriquez, J.; Isella, C.; Comoglio, P.M.; et al. IGF2 is an actionable target that identifies a distinct subpopulation of colorectal cancer patients with marginal response to anti-EGFR therapies. Sci. Transl. Med. 2015, 7, 272ra12. [Google Scholar] [CrossRef]
- Arqués, O.; Chicote, I.; Puig, I.; Tenbaum, S.P.; Argilés, G.; Dienstmann, R.; Fernández, N.; Caratù, G.; Matito, J.; Silberschmidt, D.; et al. Tankyrase Inhibition Blocks Wnt/β-Catenin Pathway and Reverts Resistance to PI3K and AKT Inhibitors in the Treatment of Colorectal Cancer. Clin. Cancer Res. 2016, 22, 644–656. [Google Scholar] [CrossRef]
- Lau, T.; Chan, E.; Callow, M.; Waaler, J.; Boggs, J.; Blake, R.A.; Magnuson, S.; Sambrone, A.; Schutten, M.; Firestein, R.; et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–3144. [Google Scholar] [CrossRef]
- Isella, C.; Brundu, F.; Bellomo, S.E.; Galimi, F.; Zanella, E.; Porporato, R.; Petti, C.; Fiori, A.; Orzan, F.; Senetta, R.; et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat. Comm. 2017, 8, 15107. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Pantel, K. Circulating tumor cells: Liquid biopsy ofcancer. Clin. Chem. 2013, 59, 110–118. [Google Scholar] [CrossRef]
- Jia, S.; Zhang, R.; Li, Z.; Li, J. Clinical and biological significance of circulating tumor cells, circulating tumor DNA, and exosomes as biomarkers in colorectal cancer. Oncotarget 2017, 8, 55632–55645. [Google Scholar] [CrossRef]
- Toiyama, Y.; Okugawa, Y.; Fleshman, J.; Richard Boland, C.; Goel, A. Micrornas as potential liquid biopsy biomarkers in colorectal cancer: A. systematic review. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 274–282. [Google Scholar] [CrossRef]
- Dominguez-Vigil, I.G.; Moreno-Martinez, A.K.; Wang, J.Y.; Roehrl, M.H.A.; Barrera-Saldana, H.A. The dawn of the liquid biopsy in the fight against cancer. Oncotarget 2018, 9, 2912–2922. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Diaz, L.A.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Earlyand Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Winther-Larsen, A.; Demuth, C.; Fledelius, J.; Madsen, A.T.; Hjorthaug, K.; Meldgaard, P.; Sorensen, B.S. Correlation between circulating mutant DNA and metabolic tumour burden in advanced non–small cell lung cancer patients. Br. J. Cancer 2017, 117, 704–709. [Google Scholar] [CrossRef]
- Thierry, A.R.; Mouliere, F.; Gongora, C.; Ollier, J.; Robert, B.; Ychou, M.; Rio, M.D.; Molina, F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010, 38, 6159–6175. [Google Scholar] [CrossRef]
- Diaz, L.A.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance totargeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Bedin, C.; Enzo, M.V.; Del Bianco, P.; Pucciarelli, S.; Nitti, D.; Agostini, M. Diagnostic and prognostic role of cell-free DNA testing for colorectal cancer patients. Int. J. Cancer 2017, 140, 1888–1898. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage ii colon cancer. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef]
- Huang, M.Y.; Tsai, H.L.; Huang, J.J.; Wang, J.Y. Clinical Implications and Future Perspectives of Circulating Tumor Cells and Biomarkers in Clinical Outcomes of Colorectal Cancer. Transl. Oncol. 2016, 9, 340–347. [Google Scholar] [CrossRef]
- Burz, C.; Pop, V.V.; Buiga, R.; Daniel, S.; Samasca, G.; Aldea, C.; Lupan, I. Circulating tumor cells in clinical research and monitoring patients with colorectal cancer. Oncotarget 2018, 9, 24561–24571. [Google Scholar] [CrossRef]
- Plaks, V.; Koopman, C.D.; Werb, Z. Cancer. Circulating tumor cells. Science 2013, 341, 1186–1188. [Google Scholar] [CrossRef]
- Bunger, S.; Zimmermann, M.; Habermann, J.K. Diversity of assessing circulating tumor cells (CTCs) emphasizes need for standardization: A CTC Guide to design and report trials. Cancer Metastasis Rev. 2015, 34, 527–545. [Google Scholar] [CrossRef]
- Chen, Y.F.; Wang, J.Y.; Wu, C.H.; Chen, F.M.; Cheng, T.L.; Lin, S.R. Detection of circulating cancer cells with K-ras oncogene using membrane array. Cancer Lett. 2005, 229, 115–122. [Google Scholar] [CrossRef]
- Steinert, G.; Scholch, S.; Koch, M.; Weitz, J. Biology and significance of circulating and disseminated tumour cells in colorectal cancer. Langenbeck Arch. Surg. 2012, 397, 535–542. [Google Scholar] [CrossRef]
- Hardingham, J.E.; Grover, P.; Winter, M.; Hewett, P.J.; Price, T.J.; Thierry, B. Detection and Clinical Significance of Circulating Tumor Cells in Colorectal Cancer—20 Years of Progress. Mol. Med. 2015, 21, 25–31. [Google Scholar] [CrossRef]
- Greening, D.W.; Gopal, S.K.; Mathias, R.A.; Liu, L.; Sheng, J.; Zhu, H.J.; Simpson, R.J. Emerging roles of exosomes during epithelial-mesenchymal transition and cancer progression. Semin. Cell Dev. Biol. 2015, 40, 60–71. [Google Scholar] [CrossRef]
- Mu, W.; Rana, S.; Zöller, M. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia 2013, 15, 875–887. [Google Scholar] [CrossRef]
- Wang, X.; Ding, X.; Nan, L.; Wang, Y.; Wang, J.; Yan, Z.; Zhang, W.; Sun, J.; Zhu, W.; Ni, B.; et al. Investigation of the roles of exosomes in colorectal cancer liver metastasis. Oncol. Rep. 2015, 33, 2445–2453. [Google Scholar] [CrossRef]
- Menéndez, P.; Villarejo, P.; Padilla, D.; Menéndez, J.M.; Montes, J.A.R. Diagnostic and prognostic significance of serum microRNAs in colorectal cancer. J. Surg. Oncol. 2013, 107, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Köberle, V.; Pleli, T.; Schmithals, C.; Alonso, E.A.; Haupenthal, J.; Bönig, H.; Peveling-Oberhag, J.; Biondi, R.M.; Zeuzem, S.; Kronenberger, B.; et al. Differential stability of cell-free circulating microRNAs: Implications for their utilization as biomarkers. PLoS ONE 2013, 8, e75184. [Google Scholar] [CrossRef] [PubMed]
- Ogata-Kawata, H.; Izumiya, M.; Kurioka, D.; Honma, Y.; Yamada, Y.; Furuta, K.; Gunji, T.; Ohta, H.; Okamoto, H.; Sonoda, H.; et al. Circulating exosomal microRNAs as biomarkers of colon cancer. PLoS ONE 2014, 9, e92921. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kobunai, T.; Yamamoto, Y.; Matsuda, K.; Ishihara, S.; Nozawa, K.; Yamada, H.; Hayama, T.; Inoue, E.; Tamura, J.; et al. Chromosomal instability (CIN) phenotype, CIN high or CIN low, predicts survival for colorectal cancer. J. Clin. Oncol. 2012, 30, 2256–2264. [Google Scholar] [CrossRef]
- Mojarad, E.N.; Kuppen, P.J.K.; Aghdaei, H.A.; Reza Zali, M. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed Bench 2013, 6, 120–128. [Google Scholar]


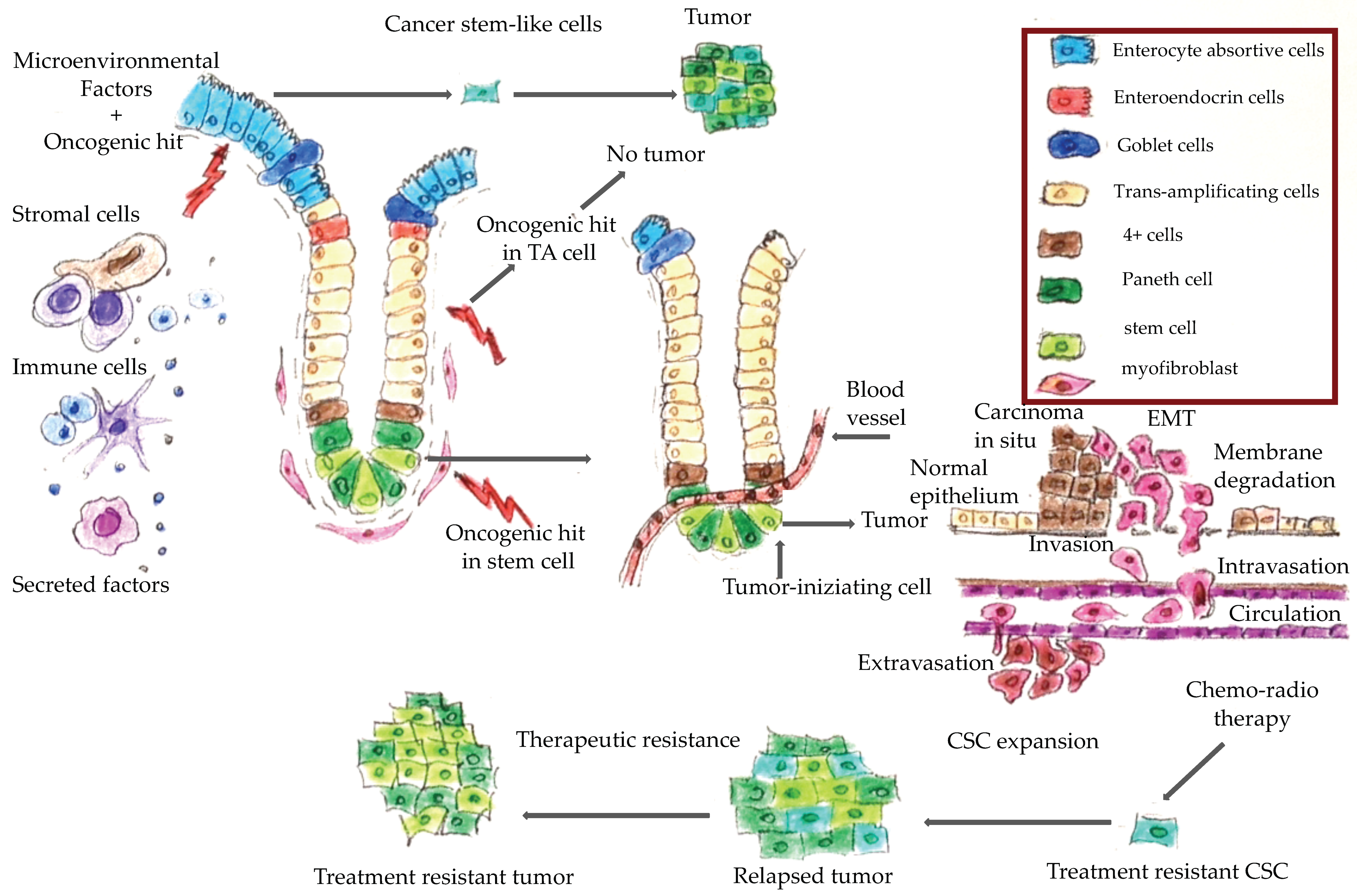{kind=link}
| Syndrome | Genes | Inheritance | Recomended Age of Screening | Tumor Molecular Features |
|---|---|---|---|---|
| Adenomatous Polyposis Syndromes | ||||
| FAP/AFAP | APC | Autosomal dominant | 20/10–12 years | CIN, APC mutations [58]. |
| PPAP | POLE, POLD1 | Autosomal dominant | none | Controversial percentage of G > T/C > A transversions [59]. |
| MAP | MUTYH | Autosomal recessive | 30–50 years [60] | KRAS, p53, APC mutations [57]. |
| NAP | NTHL1 [61] | Autosomal recessive | none | none relevant |
| MSH3 polyposis | MSH3 | Autosomal recessive | none | EMAST, MSI-L [62]. |
| Amartomatous Polyposis Syndromes | ||||
| PJS | STK11 | Autosomal dominant | 10–15 years [60] | none relevant |
| PHTS | PTEN | Autosomal dominant | none | none relevant |
| JPS | BMPR1A, SMAD4 | Autosomal dominant | 15 years or earlier [63] | none relevant |
| Mixed Polyposis | ||||
| HMPS | GREM1, BRAF | Autosomal dominant | none | BRAF and KRAS mutations, MSI [64]. |
| Serrated Adenomas | ||||
| SPS | RNF43 | Autosomal dominant | none | BRAF V600E and KRAS (codons 12 and 13) mutations, MLH1 methylation, MGMT methylation, CIMP [65]. |
| Nonpolyposis CRC | ||||
| LYNCH | MSH2, MLH1, MSH6, MSH3, PMS2, EPCAM | Autosomal dominant | 20–25 (ten years earlier than the youngest age of colon cancer diagnosis in the family) | MSI-H, MSI-L, EMAST V600E BRAF wt [66]. |
| NONPOLYPOSIS CRC-MSS | RPS20 | Autosomal dominant | none | MSI-BRAF mutations LINE-1 methylation, V600E BRAF wt [67]. |
| Biomarkers | Diagnostic Value | Prognostic Value | Predictive Value |
|---|---|---|---|
| CIN phenotype | APC mutated sporadic and hereditary CRC | marker of poor prognosis | Identify high-risk patients with stage II CRC who might benefit from adjuvant chemotherapy [9,154] |
| CIMP | Specific of serrated adenomas | Marker of poor prognosis | conflicting data exsist [155] |
| MSI | Lynch syndrome [14] Sporadic MSI tumor in combination with BRAF V600E mutation | MSI-H is associated with better prognosis and survival versus MSI-L and MSS [112] | MSI-H is associated with worse response to 5-Flurouracil-based chemotherapy compared to MSI-L and MSS [112]; dMMR–MSI-H is associated with good renponse to immunotherapy [107]. |
| BRAF V600E mutation | Sporadic MSI CRC [27]; serrated polyposis syndrome [63] | none suggested | none suggested |
| KRAS mutation | none suggested | marker of poor prognosis. | Identify patients resistant to anti-EGFR antibody treatment [82]. |
| VEGF | none suggested | marker of poor prognosis | |
| TAMs | none suggested | marker of good prognosis [69] | Identify patients who can benefit from treatment with 5-FU [91,92,93,94,95,96]. |
| TILs | none suggested | marker of good prognosis and survival [100] | Identify patients who can benefit from immunotherapy [96,101,105]. |
| CAFs | none suggested | marker of tumor and aggressivenes and poor prognosis in untreated CRC | none suggested |
| TS protein and TYMS gene expression | none suggested | High TS and TYMS expression correlates with good overall survival after chemotherapy | High TS and TYMS expression are associated with good response to 5-FU [76,77]. |
| TP protein | none suggested | none suggested | High TP expression is associated with good response to capecitabine; loss of TP function causes capecitabine-resistance [76,77]. |
| CMS1 (MSI-Immune) | none suggested | none suggested | Identify patients with poor progression-free and overall survival after EGFR treatment [115]; good response to 5-FU treatment is suggested by in vitro study [116]. |
| CMS2 (canonical subtype) | none suggested | none suggested | Identify patients with poor progression-free and overall survival after EGFR treatment [115]; good response to 5-FU treatment is suggested by in vitro study [116]; Identify patients with the best responce to oxaliplatin [117]. |
| CMS3 (metabolic subtype) | none suggested | none suggested | good response to 5-FU treatment is suggested by in vitro study [116]. |
| CMS4 (mesenchymal subtype) | none suggested | none suggested | Identify patients with better progression-free and overall survival with an irinotecan regimen than with oxaliplatin chemotherapy [115]; poor or absent responce to oxaliplatin [117]. |
| ctDNA | Allows identification of genotypic changes that occur during systemic treatment [136] | Marker of poor survival [139,140] | serial ctDNA measurements could be an early predictor of treatment response [139,140]. |
| CTCs | Marker of both early stage and metastatic cancer [145] | Marker of worse clinical outcome parameters, overall survival and progression-free survival [141,147,148] | none suggested |
| Circulating exosomal miRNAs | Marker of early detection [153] | none suggested | none suggested |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turano, M.; Delrio, P.; Rega, D.; Cammarota, F.; Polverino, A.; Duraturo, F.; Izzo, P.; De Rosa, M. Promising Colorectal Cancer Biomarkers for Precision Prevention and Therapy. Cancers 2019, 11, 1932. https://doi.org/10.3390/cancers11121932
Turano M, Delrio P, Rega D, Cammarota F, Polverino A, Duraturo F, Izzo P, De Rosa M. Promising Colorectal Cancer Biomarkers for Precision Prevention and Therapy. Cancers. 2019; 11(12):1932. https://doi.org/10.3390/cancers11121932
Chicago/Turabian StyleTurano, Mimmo, Paolo Delrio, Daniela Rega, Francesca Cammarota, Alessia Polverino, Francesca Duraturo, Paola Izzo, and Marina De Rosa. 2019. "Promising Colorectal Cancer Biomarkers for Precision Prevention and Therapy" Cancers 11, no. 12: 1932. https://doi.org/10.3390/cancers11121932
APA StyleTurano, M., Delrio, P., Rega, D., Cammarota, F., Polverino, A., Duraturo, F., Izzo, P., & De Rosa, M. (2019). Promising Colorectal Cancer Biomarkers for Precision Prevention and Therapy. Cancers, 11(12), 1932. https://doi.org/10.3390/cancers11121932