Direct Targeting Options for STAT3 and STAT5 in Cancer

 ,
,  , , ,
, , , 
Abstract
1. Introduction
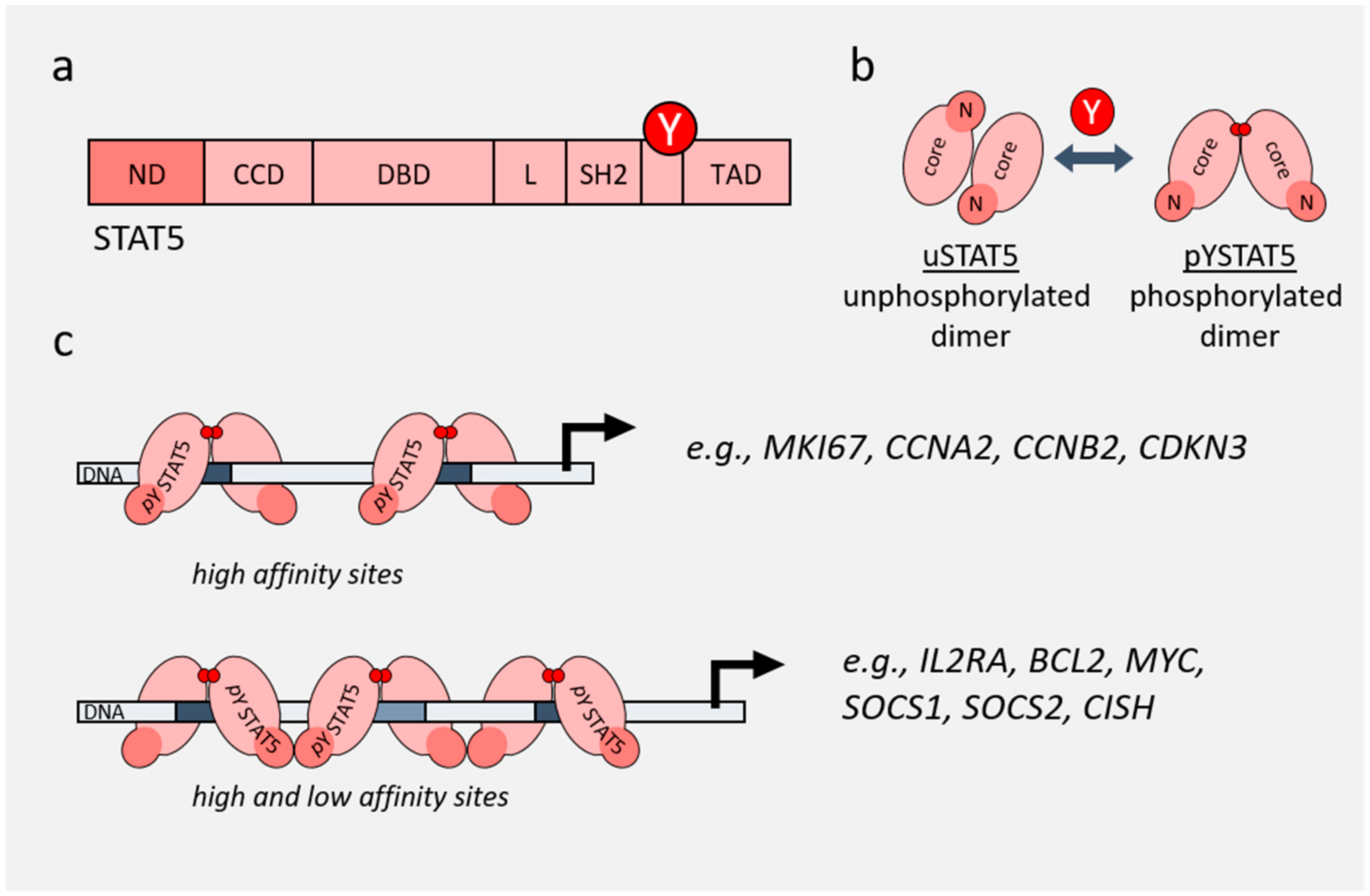
2. Role of STAT3 and STAT5 in Cancer
3. Non-Canonical Functions of STAT3 and STAT5
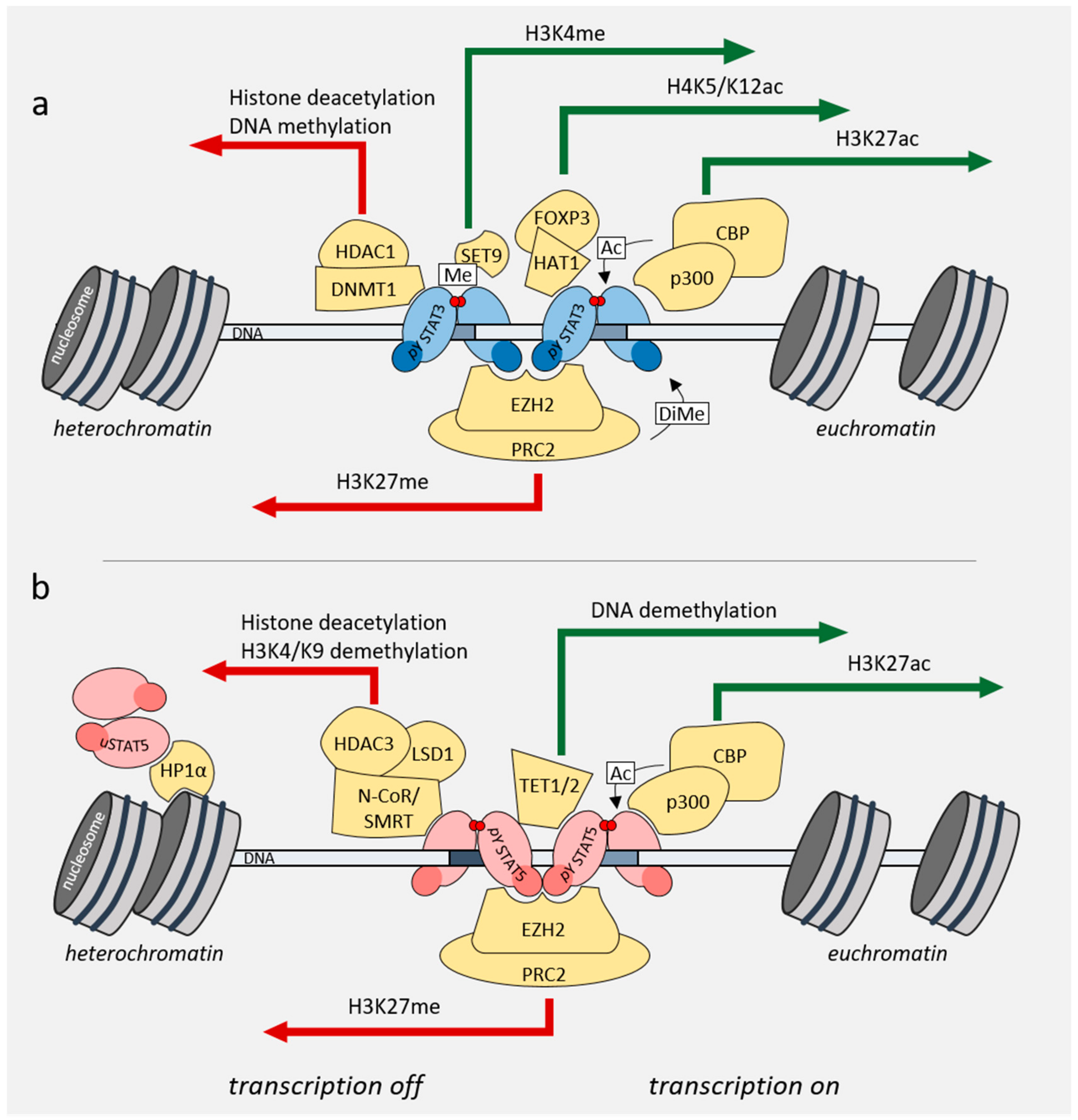
4. Role of STAT3/5 in Chromatin Landscape
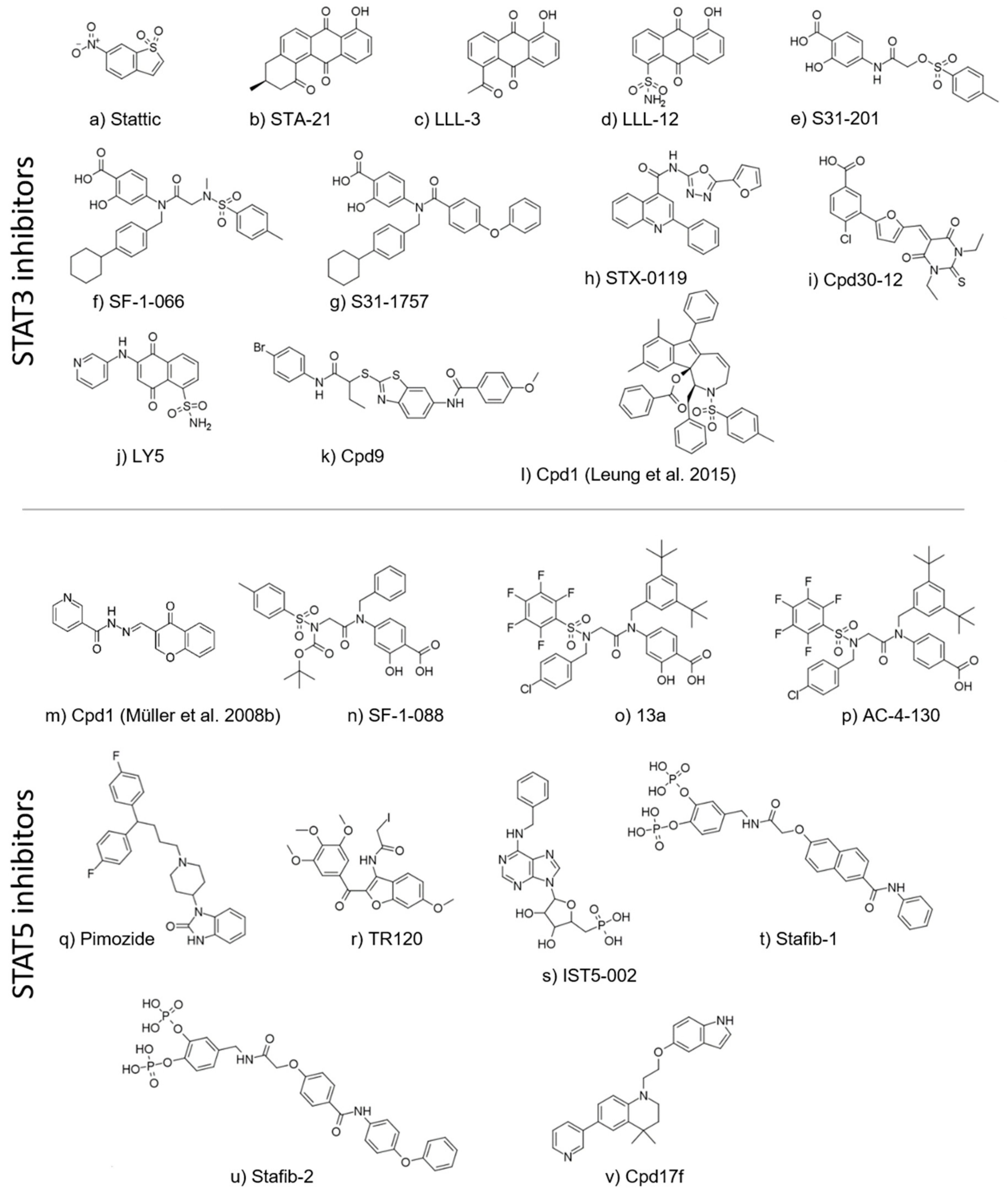
5. Direct STAT Targeting of the SH2 Domain
5.1. STAT3 Inhibitors
5.2. STAT5 Inhibitors
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Koromilas, A.E.; Sexl, V. The tumor suppressor function of STAT1 in breast cancer. JAK-STAT 2013, 2, e23353. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jove, R. The stats of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Meissl, K.; Macho-Maschler, S.; Müller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef]
- Klover, P.J.; Muller, W.J.; Robinson, G.W.; Pfeiffer, R.M.; Yamaji, D.; Hennighausen, L. Loss of STAT1 from Mouse Mammary Epithelium Results in an Increased Neu-Induced Tumor Burden. Neoplasia 2010, 12, 899–905. [Google Scholar] [CrossRef]
- Kovacic, B.; Stoiber, D.; Moriggl, R.; Weisz, E.; Ott, R.G.; Kreibich, R.; Levy, D.E.; Beug, H.; Freissmuth, M.; Sexl, V. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell 2006, 10, 77–87. [Google Scholar] [CrossRef]
- Godfrey, A.L.; Chen, E.; Massie, C.E.; Silber, Y.; Pagano, F.; Bellosillo, B.; Guglielmelli, P.; Harrison, C.N.; Reilly, J.T.; Stegelmann, F.; et al. STAT1 activation in association with JAK2 exon 12 mutations. Haematologica 2016, 101, e15. [Google Scholar] [CrossRef]
- Prutsch, N.; Gurnhofer, E.; Suske, T.; Liang, H.C.; Schlederer, M.; Roos, S.; Wu, L.C.; Simonitsch-Klupp, I.; Alvarez-Hernandez, A.; Kornauth, C.; et al. Dependency on the TYK2/STAT1/MCL1 axis in anaplastic large cell lymphoma. Leukemia 2019, 33, 696–709. [Google Scholar] [CrossRef]
- Sanda, T.; Tyner, J.W.; Gutierrez, A.; Ngo, V.N.; Glover, J.; Chang, B.H.; Yost, A.; Ma, W.; Fleischman, A.G.; Zhou, W.; et al. TYK2–STAT1–BCL2 Pathway Dependence in T-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2013, 3, 564. [Google Scholar] [CrossRef]
- Furth, P.A. STAT signaling in different breast cancer sub-types. Mol. Cell. Endocrinol. 2014, 382, 612–615. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E. STATs: Transcriptional control and biological impact: Signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an Oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Corvinus, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufman, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent STAT3 Activation in Colon Cancer Is Associated with Enhanced Cell Proliferation and Tumor Growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736. [Google Scholar] [CrossRef]
- Mohanty, S.K.; Yagiz, K.; Pradhan, D.; Luthringer, D.J.; Amin, M.B.; Alkan, S.; Cinar, B. STAT3 and STAT5A are potential therapeutic targets in castration-resistant prostate cancer. Oncotarget 2017, 8. [Google Scholar] [CrossRef]
- Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M.P.; Addya, S.; Fortina, P.; Dasgupta, A.; Hyslop, T.; et al. Transcription Factor Stat3 Stimulates Metastatic Behavior of Human Prostate Cancer Cells in Vivo, whereas Stat5b Has a Preferential Role in the Promotion of Prostate Cancer Cell Viability and Tumor Growth. Am. J. Pathol. 2010, 176, 1959–1972. [Google Scholar] [CrossRef]
- Boutillon, F.; Pigat, N.; Sackmann Sala, L.; Reyes-Gomez, E.; Moriggl, R.; Guidotti, J.-E.; Goffin, V. STAT5a/b Deficiency Delays, but does not Prevent, Prolactin-Driven Prostate Tumorigenesis in Mice. Cancers 2019, 11, 929. [Google Scholar] [CrossRef]
- Cahu, X.; Constantinescu, S.N. Oncogenic Drivers in Myeloproliferative Neoplasms: From JAK2 to Calreticulin Mutations. Curr. Hematol. Malig. Rep. 2015, 10, 335–343. [Google Scholar] [CrossRef]
- Staerk, J.; Constantinescu, S.N. The JAK-STAT pathway and hematopoietic stem cells from the JAK2 V617F perspective. JAK-STAT 2012, 1, 184–190. [Google Scholar] [CrossRef]
- Warsch, W.; Walz, C.; Sexl, V. JAK of all trades: JAK2-STAT5 as novel therapeutic targets in BCR-ABL1+ chronic myeloid leukemia. Blood 2014, 122, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Hoelbl, A.; Kovacic, B.; Kerenyi, M.A.; Simma, O.; Warsch, W.; Cui, Y.; Beug, H.; Hennighausen, L.; Moriggl, R.; Sexl, V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 2006, 107, 4898. [Google Scholar] [CrossRef]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M.; et al. STAT5BN642H is a driver mutation for T cell neoplasia. J. Clin. Invest. 2018, 128, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu. Rev. Immunol. 2017, 35, 533–550. [Google Scholar] [CrossRef]
- Schrader, A.; Crispatzu, G.; Oberbeck, S.; Mayer, P.; Pützer, S.; von Jan, J.; Vasyutina, E.; Warner, K.; Weit, N.; Pflug, N.; et al. Actionable perturbations of damage responses by TCL1/ATM and epigenetic lesions form the basis of T-PLL. Nat. Commun. 2018, 9, 697. [Google Scholar] [CrossRef]
- Koppikar, P.; Bhagwat, N.; Kilpivaara, O.; Manshouri, T.; Adli, M.; Hricik, T.; Liu, F.; Saunders, L.M.; Mullally, A.; Abdel-Wahab, O.; et al. Heterodimeric JAK–STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012, 489, 155–159. [Google Scholar] [CrossRef]
- Berger, A.; Sexl, V.; Valent, P.; Moriggl, R. Inhibition of STAT5: a therapeutic option in BCR-ABL1-driven leukemia. Oncotarget 2014, 5, 9564–9576. [Google Scholar] [CrossRef]
- Cumaraswamy, A.A.; Lewis, A.M.; Geletu, M.; Todic, A.; Diaz, D.B.; Cheng, X.R.; Brown, C.E.; Laister, R.C.; Muench, D.; Kerman, K.; et al. Nanomolar-Potency Small Molecule Inhibitor of STAT5 Protein. ACS Med. Chem. Lett. 2014, 5, 1202–1206. [Google Scholar] [CrossRef]
- Mertens, C.; Zhong, M.; Krishnaraj, R.; Zou, W.; Chen, X.; Darnell, J.E. Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev. 2006, 20, 3372–3381. [Google Scholar]
- Neculai, D.; Neculai, A.M.; Verrier, S.; Straub, K.; Klumpp, K.; Pfitzner, E.; Becker, S. Structure of the Unphosphorylated STAT5a Dimer. J. Biol. Chem. 2005, 280, 40782–40787. [Google Scholar] [CrossRef]
- Fahrenkamp, D.; Li, J.; Ernst, S.; Schmitz-Van de Leur, H.; Chatain, N.; Küster, A.; Koschmieder, S.; Lüscher, B.; Rossetti, G.; Müller-Newen, G. Intramolecular hydrophobic interactions are critical mediators of STAT5 dimerization. Sci. Rep. 2016, 6, 35454. [Google Scholar] [CrossRef] [PubMed]
- Moriggl, R.; Sexl, V.; Kenner, L.; Duntsch, C.; Stangl, K.; Gingras, S.; Hoffmeyer, A.; Bauer, A.; Piekorz, R.; Wang, D.; et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell 2005, 7, 87–99. [Google Scholar] [CrossRef]
- Lin, J.-X.; Du, N.; Li, P.; Kazemian, M.; Gebregiorgis, T.; Spolski, R.; Leonard, W.J. Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells. Nat. Commun. 2017, 8, 1320. [Google Scholar] [CrossRef]
- de Araujo, E.D.; Erdogan, F.; Neubauer, H.A.; Meneksedag-Erol, D.; Manaswiyoungkul, P.; Eram, M.S.; Seo, H.-S.; Qadree, A.K.; Israelian, J.; Orlova, A.; et al. Structural and functional consequences of the STAT5BN642H driver mutation. Nat. Commun. 2019, 10, 2517. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Li, J.; Hannah, R.; Biddie, S.; Leal-Cervantes, A.I.; Kirschner, K.; Flores Santa Cruz, D.; Sexl, V.; Go ttgens, B.; Green, A.R. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 2016, 35, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zeng, C.; Tarasova, N.I.; Chasovskikh, S.; Dritschilo, A.; Timofeeva, O.A. A new role for STAT3 as a regulator of chromatin topology. Transcription 2013, 4, 227–231. [Google Scholar] [CrossRef]
- Kornfeld, J.-W.; Grebien, F.; Kerenyi, M.A.; Friedbichler, K.; Kovacic, B.; Zankl, B.; Hoelbl, A.; Nivarti, H.; Beug, H.; Sexl, V.; et al. The different functions of Stat5 and chromatin alteration through Stat5 proteins. Front. Biosci. J. Virtual Libr. 2008, 13, 6237–6254. [Google Scholar] [CrossRef]
- Li, G.; Wang, Z.; Zhang, Y.; Kang, Z.; Haviernikova, E.; Cui, Y.; Hennighausen, L.; Moriggl, R.; Wang, D.; Tse, W.; et al. STAT5 requires the N-domain to maintain hematopoietic stem cell repopulating function and appropriate lymphoid-myeloid lineage output. Exp. Hematol. 2007, 35, 1684–1694. [Google Scholar] [CrossRef]
- Murphy, T.; Yee, K.W.L. Cytarabine and daunorubicin for the treatment of acute myeloid leukemia. Expert Opin. Pharmacother. 2017, 18, 1765–1780. [Google Scholar] [CrossRef]
- Ota, N.; Brett, T.J.; Murphy, T.L.; Fremont, D.H.; Murphy, K.M. N-domain–dependent nonphosphorylated STAT4 dimers required for cytokine-driven activation. Nat. Immunol. 2004, 5, 208–215. [Google Scholar] [CrossRef]
- Vogt, M.; Domoszlai, T.; Kleshchanok, D.; Lehmann, S.; Schmitt, A.; Poli, V.; Richtering, W.; Muller-Newen, G. The role of the N-terminal domain in dimerization and nucleocytoplasmic shuttling of latent STAT3. J. Cell Sci. 2011, 124, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Darnell, J.E. Functional Importance of Stat3 Tetramerization in Activation of the α2-Macroglobulin Gene. J. Biol. Chem. 2001, 276, 33576–33581. [Google Scholar] [CrossRef]
- Hu, T.; Yeh, J.E.; Pinello, L.; Jacob, J.; Chakravarthy, S.; Yuan, G.-C.; Chopra, R.; Frank, D.A. Impact of the N-Terminal Domain of STAT3 in STAT3-Dependent Transcriptional Activity. Mol. Cell. Biol. 2015, 35, 3284. [Google Scholar] [CrossRef]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel Roles of Unphosphorylated STAT3 in Oncogenesis and Transcriptional Regulation. Cancer Res. 2005, 65, 939. [Google Scholar] [PubMed]
- Pfeffer, S.R.; Fan, M.; Du, Z.; Yang, C.H.; Pfeffer, L.M. Unphosphorylated STAT3 regulates the antiproliferative, antiviral, and gene-inducing actions of type I interferons. Biochem. Biophys. Res. Commun. 2017, 490, 739–745. [Google Scholar] [CrossRef]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar]
- Timofeeva, O.A.; Tarasova, N.I.; Zhang, X.; Chasovskikh, S.; Cheema, A.K.; Wang, H.; Brown, M.L.; Dritschilo, A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. USA 2013, 110, 1267–1272. [Google Scholar] [CrossRef]
- Lee, J.E.; Yang, Y.-M.; Liang, F.-X.; Gough, D.J.; Levy, D.E.; Sehgal, P.B. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am. J. Physiol. Cell Physiol. 2011, 302, C804–C820. [Google Scholar] [CrossRef]
- Hu, X.; Dutta, P.; Tsurumi, A.; Li, J.; Wang, J.; Land, H.; Li, W.X. Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 10213–10218. [Google Scholar] [CrossRef]
- Engblom, D.; Kornfeld, J.-W.; Schwake, L.; Tronche, F.; Reimann, A.; Beug, H.; Hennighausen, L.; Moriggl, R.; Schutz, G. Direct glucocorticoid receptor-Stat5 interaction in hepatocytes controls body size and maturation-related gene expression. Genes Dev. 2007, 21, 1157–1162. [Google Scholar]
- Freund, P.; Kerenyi, M.A.; Hager, M.; Wagner, T.; Wingelhofer, B.; Pham, H.T.T.; Elabd, M.; Han, X.; Valent, P.; Gouilleux, F.; et al. O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies. Leukemia 2017, 31, 2132–2142. [Google Scholar] [CrossRef]
- Lin, J.-X.; Li, P.; Liu, D.; Jin, H.T.; He, J.; Rasheed, M.A.U.; Rochman, Y.; Wang, L.; Cui, K.; Liu, C.; et al. Critical Role of STAT5 Transcription Factor Tetramerization for Cytokine Responses and Normal Immune Function. Immunity 2012, 36, 586–599. [Google Scholar] [CrossRef]
- Rauth, M.; Freund, P.; Orlova, A.; Grünert, S.; Tasic, N.; Han, X.; Ruan, H.-B.; Neubauer, A.H.; Moriggl, R. Cell Metabolism Control Through O-GlcNAcylation of STAT5: A Full or Empty Fuel Tank Makes a Big Difference for Cancer Cell Growth and Survival. Int. J. Mol. Sci. 2019, 20, 1028. [Google Scholar] [CrossRef] [PubMed]
- Litterst, C.M.; Kliem, S.; Marilley, D.; Pfitzner, E. NCoA-1/SRC-1 Is an Essential Coactivator of STAT5 That Binds to the FDL Motif in the α-Helical Region of the STAT5 Transactivation Domain. J. Biol. Chem. 2003, 278, 45340–45351. [Google Scholar] [CrossRef]
- Nanou, A.; Toumpeki, C.; Lavigne, M.D.; Lazou, V.; Demmers, J.; Paparountas, T.; Thanos, D.; Katsantoni, E. The dual role of LSD1 and HDAC3 in STAT5-dependent transcription is determined by protein interactions, binding affinities, motifs and genomic positions. Nucleic Acids Res. 2016, 45, 142–154. [Google Scholar] [CrossRef]
- Yang, R.; Qu, C.; Zhou, Y.; Konkel, J.E.; Shi, S.; Liu, Y.; Chen, C.; Liu, S.; Liu, D.; Chen, Y.; et al. Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 Demethylation to Drive Regulatory T Cell Differentiation and Maintain Immune Homeostasis. Immunity 2015, 43, 251–263. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Marzec, M.; Raghunath, P.N.; Nagasawa, T.; Wasik, M.A. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 6948. [Google Scholar] [CrossRef]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 Sequence-specific DNA Binding and Transcription by p300/CREB-binding Protein-mediated Acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499. [Google Scholar] [CrossRef]
- Dasgupta, M.; Dermawan, J.K.T.; Willard, B.; Stark, G.R. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci. USA 2015, 112, 3985. [Google Scholar] [CrossRef]
- Hossain, D.M.S.; Panda, A.K.; Manna, A.; Mohanty, S.; Bhattacharjee, P.; Bhattacharyya, S.; Saha, T.; Chakraborty, S.; Kar, R.K.; Das, T.; et al. FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity 2013, 39, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Powers, S.E.; Maienschein-Cline, M.; Bartom, E.T.; Hamel, K.M.; Kee, B.L.; Dinner, A.R.; Clark, M.R. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nature Immunol. 2011, 12, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Boumahdi, S.; de Sauvage, F.J. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar]
- Becker, S.; Groner, B.; Müller, C.W. Three-dimensional structure of the Stat3β homodimer bound to DNA. Nature 1998, 394, 145–151. [Google Scholar] [CrossRef]
- Song, H.; Wang, R.; Wang, S.; Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700. [Google Scholar] [CrossRef]
- Lin, L.; Hutzen, B.; Li, P.-K.; Ball, S.; Zuo, M.; DeAngelis, S.; Foust, E.; Sobo, M.; Friedman, L.; Bhasin, D.; et al. A novel small molecule, LLL12, inhibits STAT3 phosphorylation and activities and exhibits potent growth-suppressive activity in human cancer cells. Neoplasia 2010, 12, 39–50. [Google Scholar] [CrossRef]
- Fuh, B.; Sobo, M.; Cen, L.; Josiah, D.; Hutzen, B.; Cisek, K.; Bhasin, D.; Regan, N.; Lin, L.; Chan, C.; et al. LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br. J. Cancer 2009, 100, 106–112. [Google Scholar] [CrossRef]
- Fletcher, S.; Singh, J.; Zhang, X.; Yue, P.; Page, B.D.G.; Sharmeen, S.; Shahani, V.M.; Zhao, W.; Schimmer, A.D.; Turkson, J.; et al. Disruption of transcriptionally active Stat3 dimers with non-phosphorylated, salicylic acid-based small molecules: potent in vitro and tumor cell activities. Chembiochem Eur. J. Chem. Biol. 2009, 10, 1959–1964. [Google Scholar] [CrossRef]
- Urlam, M.K.; Pireddu, R.; Ge, Y.; Zhang, X.; Sun, Y.; Lawrence, H.R.; Guida, W.C.; Sebti, S.M.; Lawrence, N.J. Development of new N-Arylbenzamides as STAT3 Dimerization Inhibitors. MedChemComm 2013, 4, 932–941. [Google Scholar] [CrossRef]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.R.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Kasembeli, M.M.; Jiang, X.; Tweardy, B.J.; Tweardy, D.J. Chemical Probes that Competitively and Selectively Inhibit Stat3 Activation. PLoS ONE 2009, 4, e4783. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Masuda, Y.; Uehara, Y.; Sato, H.; Muroya, A.; Takahashi, O.; Yokotagawa, T.; Furuya, T.; Okawara, T.; Otsuka, M.; et al. Identification of a New Series of STAT3 Inhibitors by Virtual Screening. ACS Med. Chem. Lett. 2010, 1, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-S.; Kim, H.-N.; Shin, K.D.; Yoon, Y.J.; Kim, S.-J.; Han, D.C.; Kwon, B.-M. Cryptotanshinone Inhibits Constitutive Signal Transducer and Activator of Transcription 3 Function through Blocking the Dimerization in DU145 Prostate Cancer Cells. Cancer Res. 2008, 69. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Hutzen, B.; Ball, S.; Foust, E.; Sobo, M.; Deangelis, S.; Pandit, B.; Friedman, L.; Li, C.; Li, P.-K.; et al. New curcumin analogues exhibit enhanced growth-suppressive activity and inhibit AKT and signal transducer and activator of transcription 3 phosphorylation in breast and prostate cancer cells. Cancer Sci. 2009, 100, 1719–1727. [Google Scholar] [CrossRef]
- Amani, H.; Ajami, M.; Nasseri Maleki, S.; Pazoki-Toroudi, H.; Daglia, M.; Tsetegho Sokeng, A.J.; Di Lorenzo, A.; Nabavi, S.F.; Devi, K.P.; Nabavi, S.M. Targeting signal transducers and activators of transcription (STAT) in human cancer by dietary polyphenolic antioxidants. Biochimie 2017, 142, 63–79. [Google Scholar] [CrossRef]
- Li, M.; Yue, G.G.-L.; Song, L.-H.; Huang, M.-B.; Lee, J.K.-M.; Tsui, S.K.-W.; Fung, K.-P.; Tan, N.-H.; Lau, C.B.-S. Natural small molecule bigelovin suppresses orthotopic colorectal tumor growth and inhibits colorectal cancer metastasis via IL6/STAT3 pathway. Biochem. Pharmacol. 2018, 150, 191–201. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Llorach-Parés, L.; Pérez-Sánchez, A.; Micol, V.; Nonell-Canals, A.; Joven, J.; Valiente, M.; Sánchez-Martínez, M.; Bosch-Barrera, J.; et al. Silibinin is a direct inhibitor of STAT3. Food Chem. Toxicol. 2018, 116, 161–172. [Google Scholar] [CrossRef]
- Yu, W.; Xiao, H.; Lin, J.; Li, C. Discovery of Novel STAT3 Small Molecule Inhibitors via in Silico Site-Directed Fragment-Based Drug Design. J. Med. Chem. 2013, 56, 4402–4412. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, W.; Li, Y. Discovery of novel inhibitors of signal transducer and activator of transcription 3 ( STAT3 ) signaling pathway by virtual screening. Eur. J. Med. Chem. 2013, 62, 301–310. [Google Scholar] [CrossRef]
- Leung, K.; Liu, L.; Lin, S.; Lu, L.; Zhong, H.; Susanti, D.; Rao, W.; Wang, M.; Ian, W.; Chan, D.S.; et al. Discovery of a small-molecule inhibitor of STAT3 by ligand-based pharmacophore screening. Methods 2015, 71, 38–43. [Google Scholar] [CrossRef]
- Tolcher, A.; Flaherty, K.; Shapiro, G.I.; Berlin, J.; Witzig, T.; Habermann, T.; Bullock, A.; Rock, E.; Elekes, A.; Lin, C.; et al. A First-in-Human Phase I Study of OPB-111077, a Small-Molecule STAT3 and Oxidative Phosphorylation Inhibitor, in Patients with Advanced Cancers. Oncologist 2018, 23, 658–e72. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.; Kang, J.; Lim, H.Y.; Kim, J.H.; Lee, M.-A.; Lee, K.-H.; Kim, T.-Y.; Ryoo, B.-Y. Phase I Dose-Finding Study of OPB-111077, a Novel STAT3 Inhibitor, in Patients with Advanced Hepatocellular Carcinoma. Cancer Res. Treat. 2018, 51, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, L.; Jian, L.; Yu, R.; Zhao, J.; Sun, L.; Du, G.; Liu, X. Stattic inhibits RANKL-mediated osteoclastogenesis by suppressing activation of STAT3 and NF-κB pathways. Int. Immunopharmacol. 2018, 58, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Cui, C.; Dodge, C.T.; Bhayani, M.K.; Lai, S.Y. Targeting STAT3 inhibits growth and enhances radiosensitivity in head and neck squamous cell carcinoma. Oral Oncol. 2012, 48, 1220–1226. [Google Scholar] [CrossRef]
- Miyoshi, K.; Takaishi, M.; Nakajima, K.; Ikeda, M.; Kanda, T.; Tarutani, M.; Iiyama, T.; Asao, N.; DiGiovanni, J.; Sano, S. Stat3 as a Therapeutic Target for the Treatment of Psoriasis: A Clinical Feasibility Study with STA-21, a Stat3 Inhibitor. J. Investig. Dermatol. 2011, 131, 108–117. [Google Scholar] [CrossRef]
- Nie, Y.; Li, Y.; Hu, S. A novel small inhibitor, LLL12, targets STAT3 in non-small cell lung cancer in vitro and in vivo. Oncol. Lett. 2018, 16, 5349–5354. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Pireddu, R.; Yang, H.; Urlam, M.K.; Lawrence, H.R.; Guida, W.C.; Lawrence, N.J.; Sebti, S.M. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013, 73, 1922–1933. [Google Scholar] [CrossRef]
- Xiao, H.; Bid, H.K.; Jou, D.; Wu, X.; Yu, W.; Li, C.; Houghton, P.J.; Lin, J. A Novel Small Molecular STAT3 Inhibitor, LY5, Inhibits Cell Viability, Cell Migration, and Angiogenesis in Medulloblastoma Cells. J. Biol. Chem. 2015, 290, 3418–3429. [Google Scholar] [CrossRef]
- Müller, J.; Schust, J.; Berg, T. A high-throughput assay for signal transducer and activator of transcription 5b based on fluorescence polarization. Anal. Biochem. 2008, 375, 249–254. [Google Scholar]
- Müller, J.; Sperl, B.; Reindl, W.; Kiessling, A.; Berg, T. Discovery of Chromone-Based Inhibitors of the Transcription Factor STAT5. ChemBioChem 2008, 9, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Page, B.D.G.; Khoury, H.; Laister, R.C.; Fletcher, S.; Vellozo, M.; Manzoli, A.; Yue, P.; Turkson, J.; Minden, M.D.; Gunning, P.T. Small Molecule STAT5-SH2 Domain Inhibitors Exhibit Potent Antileukemia Activity. J. Med. Chem. 2012, 55, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, E.D.; Manaswiyoungkul, P.; Israelian, J.; Park, J.; Yuen, K.; Farhangi, S.; Berger-Becvar, A.; Abu-Jazar, L.; Gunning, P.T. High-throughput thermofluor-based assays for inhibitor screening of STAT SH2 domains. J. Pharm. Biomed. Anal. 2017, 143, 159–167. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, E.D.; Manaswiyoungkul, P.; Erdogan, F.; Qadree, A.K.; Sina, D.; Tin, G.; Toutah, K.; Yuen, K.; Gunning, P.T. A functional in vitro assay for screening inhibitors of STAT5B phosphorylation. J. Pharm. Biomed. Anal. 2019, 162, 60–65. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.C.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef]
- Nelson, E.A.; Walker, S.R.; Weisberg, E.; Bar-natan, M.; Barrett, R.; Gashin, L.B.; Terrell, S.; Klitgaard, J.L.; Santo, L.; Addorio, M.R.; et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2017, 117, 3421–3430. [Google Scholar] [CrossRef]
- Rondanin, R.; Simoni, D.; Romagnoli, R.; Baruchello, R.; Marchetti, P.; Costantini, C.; Fochi, S.; Padroni, G.; Grimaudo, S.; Maria, R.; et al. Inhibition of activated STAT5 in Bcr/Abl expressing leukemia cells with new pimozide derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 4568–4574. [Google Scholar]
- Romagnoli, R.; Baraldi, P.G.; Prencipe, F.; Lopez-Cara, C.; Rondanin, R.; Simoni, D.; Hamel, E.; Grimaudo, S.; Pipitone, R.M.; Meli, M.; et al. Novel iodoacetamido benzoheterocyclic derivatives with potent antileukemic activity are inhibitors of STAT5 phosphorylation. Eur. J. Med. Chem. 2016, 108, 39–52. [Google Scholar] [CrossRef]
- Liao, Z.; Gu, L.; Vergalli, J.; Mariani, S.A.; De Dominici, M.; Lokareddy, R.K.; Dagvadorj, A.; Purushottamachar, P.; McCue, P.A.; Trabulsi, E.; et al. Structure-based screen identifies a potent small-molecule inhibitor of Stat5a/b with therapeutic potential for prostate cancer and chronic myeloid leukemia. Mol. Cancer Ther. 2015, 14, 1777–1794. [Google Scholar] [CrossRef]
- Elumalai, N.; Berg, A.; Natarajan, K.; Scharow, A.; Berg, T. Nanomolar Inhibitors of the Transcription Factor STAT5b with High Selectivity over STAT5a. Angew. Chem. Int. Ed. 2015, 54, 4758–4763. [Google Scholar] [CrossRef]
- Elumalai, N.; Berg, A.; Rubner, S.; Blechschmidt, L.; Song, C.; Natarajan, K.; Matysik, J.; Berg, T. Rational development of Stafib-2: a selective, nanomolar inhibitor of the transcription factor STAT5b. Sci. Rep. 2017, 7, 819. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Müller-Klieser, D.; Rubner, S.; Berg, T. Stafia-1: a STAT5a-selective inhibitor developed via docking-based screening of in silico O-phosphorylated fragments. Chem. Eur. J. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Juen, L.; Brachet-Botineau, M.; Parmenon, C.; Bourgeais, J.; Hérault, O.; Gouilleux, F.; Viaud-Massuard, M.-C.; Prié, G. New Inhibitor Targeting Signal Transducer and Activator of Transcription 5 (STAT5) Signaling in Myeloid Leukemias. J. Med. Chem. 2017, 60, 6119–6136. [Google Scholar] [CrossRef]
- Berg, A.; Sperl, B.; Berg, T. ATP Inhibits the Transcription Factor STAT5b. ChemBioChem 2019, 20, 2227–2231. [Google Scholar] [CrossRef]
- Wong, E.L.; Nawrotzky, E.; Arkona, C.; Kim, B.G.; Beligny, S.; Wang, X.; Wagner, S.; Lisurek, M.; Carstanjen, D.; Rademann, J. The transcription factor STAT5 catalyzes Mannich ligation reactions yielding inhibitors of leukemic cell proliferation. Nat. Commun. 2019, 10, 66. [Google Scholar] [CrossRef]
- Parmenon, C.; Guillard, J.; Caignard, D.-H.; Hennuyer, N.; Staels, B.; Audinot-Bouchez, V.; Boutin, J.-A.; Dacquet, C.; Ktorza, A.; Viaud-Massuard, M.-C. 4,4-Dimethyl-1,2,3,4-tetrahydroquinoline-based PPARα/γ agonists. Part I: Synthesis and pharmacological evaluation. Bioorg. Med. Chem. Lett. 2008, 18, 1617–1622. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Cpd | Protein-Based | Cell-Based | In Vivo Application Tested | Refs. | |||
|---|---|---|---|---|---|---|---|
| Assay | IC50 or Ki [µM] | Cell Line | Readout | IC50 [µM]a | |||
| Stattic | pY binding | 5.1 | HepG2, MDA-MB-231 | viability | 3.8 | [64,84,85] | |
| RAW264.7 | pYSTAT3 | s (20) | Osteoclasto genesis in C57/BL6 mice (10 mg/kg) | ||||
| M-SCC-17B, OSC-19, Cal33, UM-SCC-22 | viability | 2.2–3.5 | Head and neck cancer xenograft (50 mg/kg) | ||||
| STA-21 | Caov-3 | reporter assay | s (20) | Psoriatic disease in mouse model and phase I in clinical trial (NCT0104794) | [66,86] | ||
| MDA-MB-435s, MDA-MB-468, MDA-MB-231 | DNA binding, viability | s (20) | |||||
| LLL-3 | U373, MDA-MB-231 | DNA binding | s (20) | Xenograft glioblastoma (50 mg/kg) | [68] | ||
| MDA-MB-231 | reporter assay | s (20) | |||||
| LLL-12 | MDA-MB-231, SK-BR-3, PANC-1, HPAC, U87, U373, A549 | viability, pYSTAT3, reporter assay | 0.16–3.09 | Glioblastoma, breast cancer xenograft (2.5, 5 mg/kg) Lung cancer xenograft (20, 10 mg/kg) | [67,87] | ||
| S31-201 | 80 | NIH 3T3/v-Src | DNA binding | 86 | Xenograft breast cancer (5 mg/kg) | [69,71] | |
| MDA-MB-468, MDA-MB-231 | pYSTAT3 | s (100) | |||||
| DU145, MDA468, OCI-AML-2 | viability | 28–112 | |||||
| SF-1-066 | FP | 20 | NIH 3T3/v-Src | DNA binding | 35 | [69] | |
| DU145, MDA468, OCI-AML-2 | viability | 17–37 | |||||
| S31-1757 | pY binding | 13.5 | HEK293 | CoIP | s (50) | [88] | |
| MDA-MB-468, A549 | pYSTAT3, reporter assay | s (50) | |||||
| STX-0119 | HeLa | reporter assay | 74 | Xenograft lymphoma (160 mg/kg) | [73] | ||
| HEK293 | FRET-based dimerization | s (50) | |||||
| Cpd30-12 | pY binding | 114 | HepG2, MEF/GFP-Stat3α, MDA-MB-468, MDA-MB-231, MBA-MD-435, MCF7 | pYSTAT3, nuclear translocation, apoptosis | 60 | [72] | |
| LY5 | FP | 2.5 | U2OS, RH30, RD2, MDA-MB-231 | viability, pYSTAT3 | 0.52–1.39 | Xenograft breast cancer (5 mg/kg) | [79,89] |
| UW426, UW288-1, DAOY | pYSTAT3 | s (0.5) | |||||
| Cpd9 | HepG2/STAT3 | reporter assay, pYSTAT3 | 3.57 | [80] | |||
| MDA-MB-468 | viability | 8.83 | |||||
| Cpd1 | FP | ~10 | HeLa | reporter assay, DNA binding | ~10 | [81] | |
| Cpd | Protein-Based | Cell-Based | In Vivo Application Tested | Refs. | |||
|---|---|---|---|---|---|---|---|
| Assay | IC50 or Ki [µM]a | Cell Line | Readout | IC50 [µM]a | |||
| Cpd1 | FP | 47 | K562, Daudi | DNA binding, pYSTAT5 | s (100) | [91] | |
| SF-1-088 | FP | 8.3 | K562,MV4-11 | viability, pYSTAT5 | 80–77 | [92] | |
| 13a | K562 | pYSTAT5, viability | s (15) | [28,93,94] | |||
| MV4-11 | viability | 3.5 | |||||
| AC-4-130 | binding (thermal shift) | s (100) | MV4-11, MOLM-13 | viability, reporter assay | 1.7–1.9 | AML xenograft (25 mg/kg) | [95] |
| AML patient samples | viability | 1.6–4.9 | |||||
| Pimozide | KU812, K562 | pYSTAT5, viability | s (5) | Approved by FDA as antipsychotic drug | [96] | ||
| TR120 | K562 | viability | 0.12 | [98] | |||
| apoptosis | 0.45 | ||||||
| IST5-002 | K562, DU145, PC-3, COS-7 | pYSTAT5, reporter assay, DNA binding | s (5) | Prostate cancer xenograft (25, 50, 100 mg/kg) | [99] | ||
| viability | 3.5 | ||||||
| Stafib-1 | FP | 0.044 | K562 | pYSTAT5 | s (3) | [100] | |
| Stafib-2 | FP | 0.009 | K562 | pYSTAT5 | 1.5 | [101] | |
| Cpd17f | K562, KU812, KG1a, MV4-11 | viability, pYSTAT5 | 2.6–22.7 | [103] | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orlova, A.; Wagner, C.; de Araujo, E.D.; Bajusz, D.; Neubauer, H.A.; Herling, M.; Gunning, P.T.; Keserű, G.M.; Moriggl, R. Direct Targeting Options for STAT3 and STAT5 in Cancer. Cancers 2019, 11, 1930. https://doi.org/10.3390/cancers11121930
Orlova A, Wagner C, de Araujo ED, Bajusz D, Neubauer HA, Herling M, Gunning PT, Keserű GM, Moriggl R. Direct Targeting Options for STAT3 and STAT5 in Cancer. Cancers. 2019; 11(12):1930. https://doi.org/10.3390/cancers11121930
Chicago/Turabian StyleOrlova, Anna, Christina Wagner, Elvin D. de Araujo, Dávid Bajusz, Heidi A. Neubauer, Marco Herling, Patrick T. Gunning, György M. Keserű, and Richard Moriggl. 2019. "Direct Targeting Options for STAT3 and STAT5 in Cancer" Cancers 11, no. 12: 1930. https://doi.org/10.3390/cancers11121930
APA StyleOrlova, A., Wagner, C., de Araujo, E. D., Bajusz, D., Neubauer, H. A., Herling, M., Gunning, P. T., Keserű, G. M., & Moriggl, R. (2019). Direct Targeting Options for STAT3 and STAT5 in Cancer. Cancers, 11(12), 1930. https://doi.org/10.3390/cancers11121930