ER Stress and Unfolded Protein Response in Cancer Cachexia



{kind=link}
{kind=link}
Abstract
1. Introduction
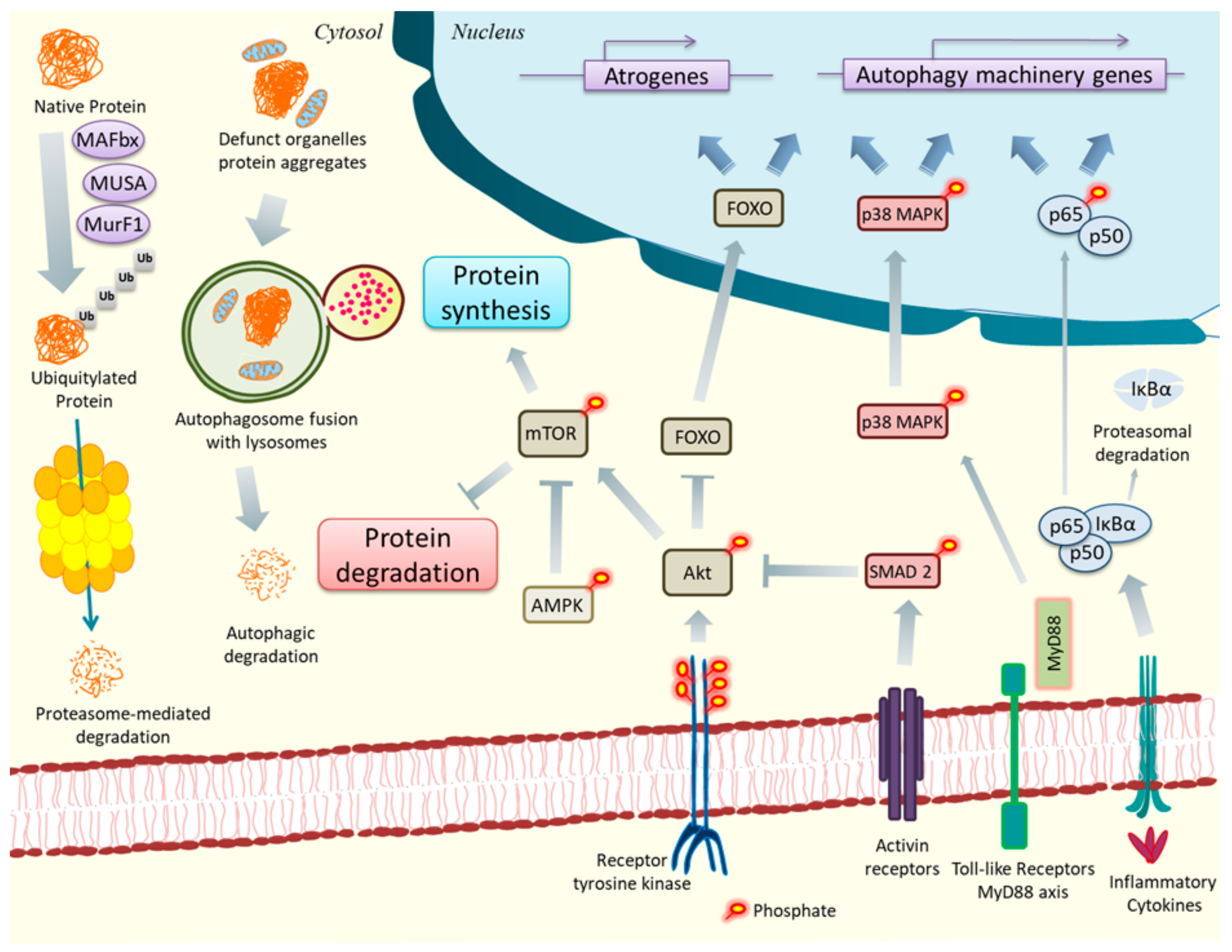
2. ER Stress, UPR, and Cancer Cachexia
3. PERK and Cachexia
4. IRE1/XBP1 and Cachexia
5. ATF6 and Cachexia
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Von Haehling, S.; Anker, S.D. Prevalence, incidence and clinical impact of cachexia: Facts and numbers-update 2014. J. CachexiaSarcopenia Muscle 2014, 5, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, A.R.; Kamal, A.H.; LeBlanc, T.W.; Ma, J.D.; Baracos, V.E.; Roeland, E.J. Cancer Cachexia: Beyond Weight Loss. J. Oncol. Pract. 2016, 12, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Houten, L.; Reilley, A.A. An investigation of the cause of death from cancer. J. Surg. Oncol. 1980, 13, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Rhee, C.; Sim, J.J.; Stenvinkel, P.; Anker, S.D.; Kovesdy, C.P. Why cachexia kills: Examining the causality of poor outcomes in wasting conditions. J. CachexiaSarcopenia Muscle 2013, 4, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Dhanapal, R.; Saraswathi, T.; Govind, R.N. Cancer cachexia. J. Oral Maxillofac. Pathol. JOMFP 2011, 15, 257. [Google Scholar] [CrossRef]
- Deans, C.; Wigmore, S.J. Systemic inflammation, cachexia and prognosis in patients with cancer. Clin. Nutr. Metab. Care 2005, 8, 265–269. [Google Scholar] [CrossRef]
- Coss, C.C.; Clinton, S.K.; Phelps, M.A. Cachectic Cancer Patients: Immune to Checkpoint Inhibitor Therapy? Clin. Cancer Res. 2018, 24, 5787–5789. [Google Scholar] [CrossRef]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef]
- Luo, Y.; Yoneda, J.; Ohmori, H.; Sasaki, T.; Shimbo, K.; Eto, S.; Kato, Y.; Miyano, H.; Kobayashi, T.; Sasahira, T.; et al. Cancer usurps skeletal muscle as an energy repository. Cancer Res. 2014, 74, 330–340. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Baccino, F.M.; Costelli, P. Coming back: Autophagy in cachexia. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis Model. Mech 2013, 6, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef]
- Sartori, R.; Gregorevic, P.; Sandri, M. TGFbeta and BMP signaling in skeletal muscle: Potential significance for muscle-related disease. Trends Endocrinol. Metab. 2014, 25, 464–471. [Google Scholar] [CrossRef]
- Lilienbaum, A. Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 2013, 4, 1–26. [Google Scholar]
- Schwalm, C.; Jamart, C.; Benoit, N.; Naslain, D.; Prémont, C.; Prevet, J.; Van Thienen, R.; Deldicque, L.; Francaux, M. Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. FASEB J. 2015, 29, 3515–3526. [Google Scholar] [CrossRef]
- Bartoli, M.; Richard, I. Calpains in muscle wasting. Int. J. Biochem. Cell Biol. 2005, 37, 2115–2133. [Google Scholar] [CrossRef]
- Sandri, M. Protein breakdown in cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 11–19. [Google Scholar] [CrossRef]
- Egerman, M.A.; Glass, D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Duval, A.P.; Jeanneret, C.; Santoro, T.; Dormond, O. mTOR and tumor cachexia. Int. J. Mol. Sci. 2018, 19, 2225. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.; Padrão, A.I.; Maciel, E.; Santinha, D.; Oliveira, P.; Vitorino, R.; Moreira-Gonçalves, D.; Colaço, B.; Pires, M.J.; Nunes, C.; et al. Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Fermoselle, C.; García-Arumí, E.; Puig-Vilanova, E.; Andreu, A.L.; Urtreger, A.J.; De Kier Joffé, E.D.B.; Tejedor, A.; Puente-Maestu, L.; Barreiro, E. Mitochondrial dysfunction and therapeutic approaches in respiratory and limb muscles of cancer cachectic mice. Exp. Physiol. 2013, 98, 1349–1365. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Marcondes, M.C.C.; Tisdale, M.J. Induction of protein catabolism and the ubiquitin-proteasome pathway by mild oxidative stress. Cancer Lett. 2002, 180, 69–74. [Google Scholar] [CrossRef]
- Mastrocola, R.; Reffo, P.; Penna, F.; Tomasinelli, C.E.; Boccuzzi, G.; Baccino, F.M.; Aragno, M.; Costelli, P. Muscle wasting in diabetic and in tumor-bearing rats: Role of oxidative stress. Free Radic. Biol. Med. 2008, 44, 584–593. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Beltrà, M.; De Lucia, S.; García Castillo, L.; Costelli, P. The Skeletal Muscle as an Active Player Against Cancer Cachexia. Front. Physiol. 2019, 10, 41. [Google Scholar] [CrossRef]
- Busquets, S.; Almendro, V.; Barreiro, E.; Figueras, M.; Argilés, J.M.; López-Soriano, F.J. Activation of UCPs gene expression in skeletal muscle can be independent on both circulating fatty acids and food intake. FEBS Lett. 2005, 579, 717–722. [Google Scholar] [CrossRef]
- Sanders, P.M.; Tisdale, M.J. Effect of zinc-α2-glycoprotein (ZAG) on expression of uncoupling proteins in skeletal muscle and adipose tissue. Cancer Lett. 2004, 212, 71–81. [Google Scholar] [CrossRef]
- Tzika, A.A.; Fontes-Oliveira, C.C.; Shestov, A.A.; Constantinou, C.; Psychogios, N.; Righi, V.; Mintzopoulos, D.; Busquets, S.; Lopez-Soriano, F.J.; Milot, S.; et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int. J. Oncol. 2013, 43, 886–894. [Google Scholar] [CrossRef]
- Purcell, S.A.; Elliott, S.A.; Baracos, V.E.; Chu, Q.S.C.; Prado, C.M. Key determinants of energy expenditure in cancer and implications for clinical practice. Eur. J. Clin. Nutr. 2016, 70, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Loumaye, A.; De Barsy, M.; Nachit, M.; Lause, P.; Frateur, L.; Van Maanen, A.; Trefois, P.; Gruson, D.; Thissen, J.P. Role of activin A and myostatin in human cancer cachexia. J. Clin. Endocrinol. Metab. 2015, 100, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, Z.; Ding, H.; Zhou, Y.; Doan, H.A.; Sin, K.W.T.; Zhu, Z.J.; Flores, R.; Wen, Y.; Gong, X.; et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun. 2017, 8, 589. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, K.R.; Goli, P.; Roy, A.; Sharma, A.K.; Xiong, G.; Gallot, Y.S.; Kumar, A. The Toll-Like Receptor/MyD88/XBP1 Signaling Axis Mediates Skeletal Muscle Wasting during Cancer Cachexia. Mol. Cell. Biol. 2019, 39, MCB-00184. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Burfeind, K.G.; Michaelis, K.A.; Braun, T.P.; Olson, B.; Pelz, K.R.; Morgan, T.K.; Marks, D.L. MyD88 signalling is critical in the development of pancreatic cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 10, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell. Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell. Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Kaufman, R.J. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006, 13, 374–384. [Google Scholar] [CrossRef]
- Isaac, S.T.; Tan, T.C.; Polly, P. Endoplasmic Reticulum Stress, Calcium Dysregulation and Altered Protein Translation: Intersection of Processes That Contribute to Cancer Cachexia Induced Skeletal Muscle Wasting. Curr Drug Targets 2016, 17, 1140–1146. [Google Scholar] [CrossRef]
- Afroze, D.; Kumar, A. ER stress in skeletal muscle remodeling and myopathies. FEBS J. 2019, 286, 379–398. [Google Scholar] [CrossRef]
- Bohnert, K.R.; McMillan, J.D.; Kumar, A. Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease. J. Cell. Physiol. 2018, 233, 67–78. [Google Scholar] [CrossRef]
- Barreiro, E.; Salazar-Degracia, A.; Sancho-Munoz, A.; Gea, J. Endoplasmic reticulum stress and unfolded protein response profile in quadriceps of sarcopenic patients with respiratory diseases. J. Cell. Physiol. 2019, 234, 11315–11329. [Google Scholar] [CrossRef]
- Paul, P.K.; Bhatnagar, S.; Mishra, V.; Srivastava, S.; Darnay, B.G.; Choi, Y.; Kumar, A. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol. Cell. Biol. 2012, 32, 1248–1259. [Google Scholar] [CrossRef]
- Paul, P.K.; Gupta, S.K.; Bhatnagar, S.; Panguluri, S.K.; Darnay, B.G.; Choi, Y.; Kumar, A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J. Cell. Biol. 2010, 191, 1395–1411. [Google Scholar] [CrossRef]
- Bohnert, K.R.; Gallot, Y.S.; Sato, S.; Xiong, G.; Hindi, S.M.; Kumar, A. Inhibition of ER stress and unfolding protein response pathways causes skeletal muscle wasting during cancer cachexia. FASEB J. 2016, 30, 3053–3068. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Zode, G.S.; Kuehn, M.H.; Nishimura, D.Y.; Searby, C.C.; Mohan, K.; Grozdanic, S.D.; Bugge, K.; Anderson, M.G.; Clark, A.F.; Stone, E.M.; et al. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J. Clin. Investig. 2015, 125, 3303. [Google Scholar] [CrossRef] [PubMed]
- Hentila, J.; Nissinen, T.A.; Korkmaz, A.; Lensu, S.; Silvennoinen, M.; Pasternack, A.; Ritvos, O.; Atalay, M.; Hulmi, J.J. Activin Receptor Ligand Blocking and Cancer Have Distinct Effects on Protein and Redox Homeostasis in Skeletal Muscle and Liver. Front. Physiol. 2018, 9, 1917. [Google Scholar] [CrossRef]
- Zhang, P.; McGrath, B.; Li, S.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, D.R. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 2002, 22, 3864–3874. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, D.; Li, Y.; Iida, K.; McGrath, B.; Cavener, D.R. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006, 4, 491–497. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2150–2163. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Peixeiro, I.; Romão, L. Gene Expression Regulation by Upstream Open Reading Frames and Human Disease. PLoS Genet. 2013, 9, e1003529. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci. Signal. 2016, 9, ra17. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Püschel, F.; León-Annicchiarico, C.L.; O’Connor, H.; Martin, S.J.; Palou-Gramón, D.; Lucendo, E.; Muñoz-Pinedo, C. Glucose Deprivation Induces ATF4-Mediated Apoptosis through TRAIL Death Receptors. Mol. Cell. Biol. 2017, 37, e00479-16. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Hindi, S.M.; Mann, A.K.; Gallot, Y.S.; Bohnert, K.R.; Cavener, D.R.; Whittemore, S.R.; Kumar, A. The PERK arm of the unfolded protein response regulates satellite cell-mediated skeletal muscle regeneration. Elife 2017, 6, e22871. [Google Scholar] [CrossRef] [PubMed]
- Zismanov, V.; Chichkov, V.; Colangelo, V.; Jamet, S.; Wang, S.; Syme, A.; Koromilas, A.E.; Crist, C. Phosphorylation of eIF2alpha Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell 2016, 18, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Kuroda, M.; Kiyonari, H.; Takehana, K.; Hisanaga, S.; Morimoto, M.; Zhang, J.; Oyadomari, M.; Sakaue, H.; Oyadomari, S. Ligand-induced rapid skeletal muscle atrophy in HSA-Fv2E-PERK transgenic mice. PLoS ONE 2017, 12, e0179955. [Google Scholar] [CrossRef]
- Miyake, M.; Nomura, A.; Ogura, A.; Takehana, K.; Kitahara, Y.; Takahara, K.; Tsugawa, K.; Miyamoto, C.; Miura, N.; Sato, R.; et al. Skeletal muscle-specific eukaryotic translation initiation factor 2α phosphorylation controls amino acid metabolism and fibroblast growth factor 21-mediated non-cell-autonomous energy metabolism. FASEB J. 2016, 30, 798–812. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, A.M.; Adachi, H.; Katsuno, M.; Sobue, G.; Yue, Z.; Robins, D.M.; Lieberman, A.P. Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet. 2011, 7, e1002321. [Google Scholar] [CrossRef]
- Gallot, Y.S.; Bohnert, K.R.; Straughn, A.R.; Xiong, G.; Hindi, S.M.; Kumar, A. PERK regulates skeletal muscle mass and contractile function in adult mice. FASEB J. 2019, 33, 1946–1962. [Google Scholar] [CrossRef]
- Ebert, S.M.; Monteys, A.M.; Fox, D.K.; Bongers, K.S.; Shields, B.E.; Malmberg, S.E.; Davidson, B.L.; Suneja, M.; Adams, C.M. The Transcription Factor ATF4 Promotes Skeletal Myofiber Atrophy during Fasting. Mol. Endocrinol. 2010, 24, 790. [Google Scholar] [CrossRef]
- Ebert, S.M.; Dyle, M.C.; Kunkel, S.D.; Bullard, S.A.; Bongers, K.S.; Fox, D.K.; Dierdorff, J.M.; Foster, E.D.; Adams, C.M. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J. Biol. Chem. 2012, 287, 27290–27301. [Google Scholar] [CrossRef] [PubMed]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell. Biol. 2001, 3, 158. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell. Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar] [CrossRef] [PubMed]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 14, 152–157. [Google Scholar]
- Zhang, K.; Wong, H.N.; Song, B.; Miller, C.N.; Scheuner, D.; Kaufman, R.J. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Investig. 2005, 115, 268–281. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Blais, A.; Tsikitis, M.; Acosta-Alvear, D.; Sharan, R.; Kluger, Y.; Dynlacht, B.D. An initial blueprint for myogenic differentiation. Genes Dev. 2005, 19, 553–569. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Martinon, F.; Chen, X.; Lee, A.-H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 2010, 21, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 1999, 339(Pt. 1), 135–141. [Google Scholar]
- Vallejo, M.; Ron, D.; Miller, C.P.; Habener, J.F. C/ATF, a member of the activating transcription factor family of DNA-binding proteins, dimerizes with CAAT/enhancer-binding proteins and directs their binding to cAMP response elements. Proc. Natl. Acad. Sci. USA 1993, 90, 4679–4683. [Google Scholar] [CrossRef]
- Nakanishi, K.; Sudo, T.; Morishima, N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J. Cell Biol. 2005, 169, 555–560. [Google Scholar] [CrossRef]
- Wu, J.; Ruas, J.L.; Estall, J.L.; Rasbach, K.A.; Choi, J.H.; Ye, L.; Boström, P.; Tyra, H.M.; Crawford, R.W.; Campbell, K.P.; et al. The Unfolded Protein Response Mediates Adaptation to Exercise in Skeletal Muscle through a PGC-1α/ATF6α Complex. Cell Metab. 2011, 13, 160–169. [Google Scholar] [CrossRef]
- Raciti, G.A.; Iadicicco, C.; Ulianich, L.; Vind, B.F.; Gaster, M.; Andreozzi, F.; Longo, M.; Teperino, R.; Ungaro, P.; Di Jeso, B.; et al. Glucosamine-induced endoplasmic reticulum stress affects GLUT4 expression via activating transcription factor 6 in rat and human skeletal muscle cells. Diabetologia 2010, 53, 955–965. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, A.; Kumar, A. ER Stress and Unfolded Protein Response in Cancer Cachexia. Cancers 2019, 11, 1929. https://doi.org/10.3390/cancers11121929
Roy A, Kumar A. ER Stress and Unfolded Protein Response in Cancer Cachexia. Cancers. 2019; 11(12):1929. https://doi.org/10.3390/cancers11121929
Chicago/Turabian StyleRoy, Anirban, and Ashok Kumar. 2019. "ER Stress and Unfolded Protein Response in Cancer Cachexia" Cancers 11, no. 12: 1929. https://doi.org/10.3390/cancers11121929
APA StyleRoy, A., & Kumar, A. (2019). ER Stress and Unfolded Protein Response in Cancer Cachexia. Cancers, 11(12), 1929. https://doi.org/10.3390/cancers11121929