How Prostate Cancer Cells Use Strategy Instead of Brute Force to Achieve Metastasis

Abstract
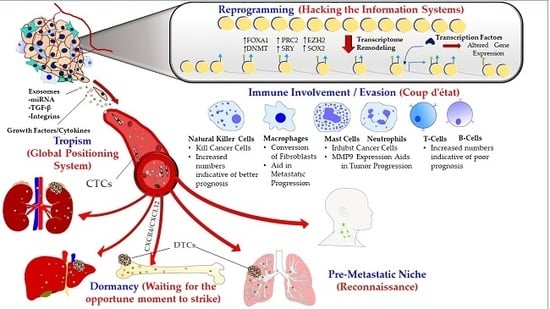
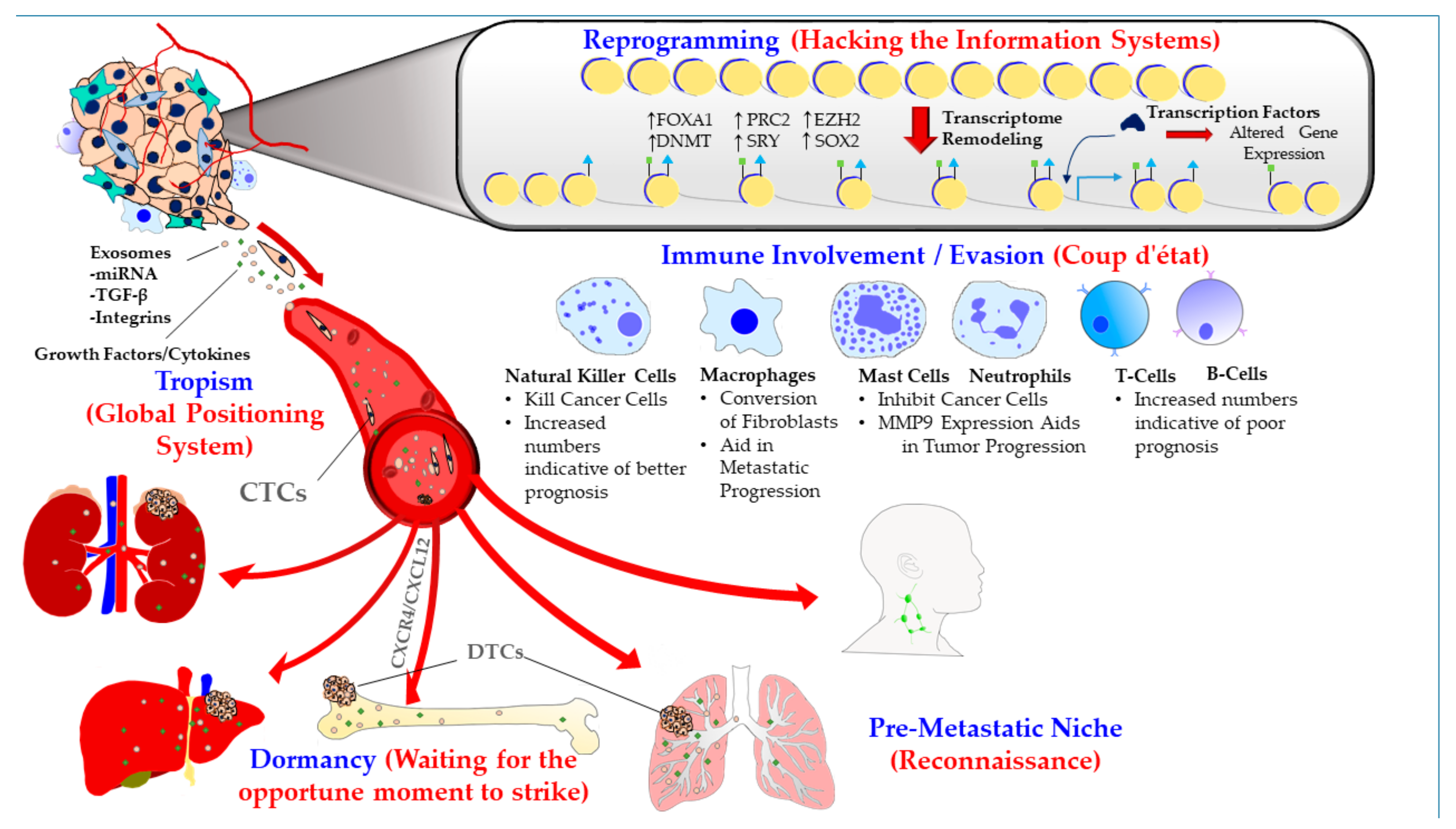
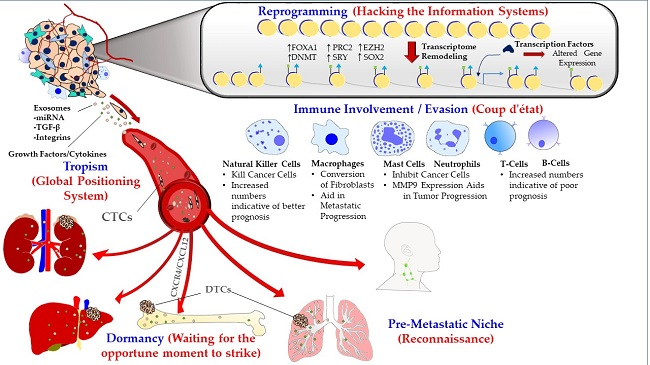{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Metastatic Tropism (Global Positioning System)
1.2. Pre-Metastatic Niche (Sending a Reconnaissance Team Ahead of the Troops)
1.3. Immune Involvement/Evasion (Coup d’état)
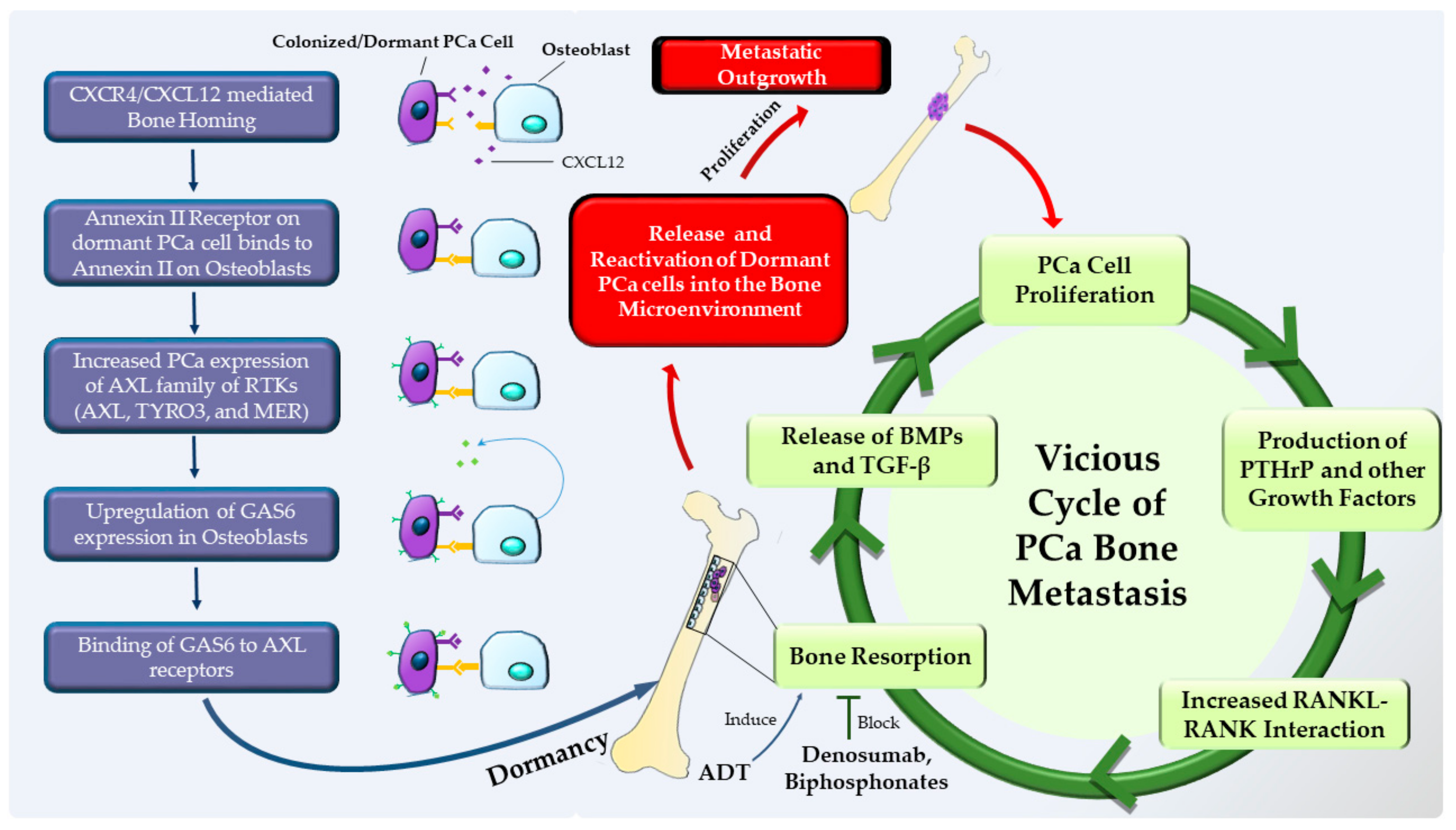
1.4. Dormancy at the Secondary Site (Staying Underground and Waiting for the Opportune Moment to Strike)
1.5. Reprogramming (Hacking the Information Systems)
2. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Cameron, M.D.; Schmidt, E.E.; Kerkvliet, N.; Nadkarni, K.V.; Morris, V.L.; Groom, A.C.; Chambers, A.F.; MacDonald, I.C. Temporal progression of metastasis in lung: Cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 2000, 60, 2541–2546. [Google Scholar] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Chinni, S.R.; Yamamoto, H.; Dong, Z.; Sabbota, A.; Bonfil, R.D.; Cher, M.L. CXCL12/CXCR4 transactivates HER2 in lipid rafts of prostate cancer cells and promotes growth of metastatic deposits in bone. Mol. Cancer Res. 2008, 6, 446–457. [Google Scholar] [CrossRef]
- Sun, Y.X.; Schneider, A.; Jung, Y.; Wang, J.; Dai, J.; Wang, J.; Cook, K.; Osman, N.I.; Koh-Paige, A.J.; Shim, H.; et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J. Bone Miner. Res. 2005, 20, 318–329. [Google Scholar] [CrossRef]
- Sun, Y.X.; Wang, J.; Shelburne, C.E.; Lopatin, D.E.; Chinnaiyan, A.M.; Rubin, M.A.; Pienta, K.J.; Taichman, R.S. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J. Cell. Biochem. 2003, 89, 462–473. [Google Scholar] [CrossRef]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar]
- Akashi, T.; Koizumi, K.; Tsuneyama, K.; Saiki, I.; Takano, Y.; Fuse, H. Chemokine receptor CXCR4 expression and prognosis in patients with metastatic prostate cancer. Cancer Sci. 2008, 99, 539–542. [Google Scholar] [CrossRef]
- Singh, A.P.; Arora, S.; Bhardwaj, A.; Srivastava, S.K.; Kadakia, M.P.; Wang, B.; Grizzle, W.E.; Owen, L.B.; Singh, S. CXCL12/CXCR4 protein signaling axis induces sonic hedgehog expression in pancreatic cancer cells via extracellular regulated kinase- and Akt kinase-mediated activation of nuclear factor kappaB: Implications for bidirectional tumor-stromal interactions. J. Biol. Chem. 2012, 287, 39115–39124. [Google Scholar] [CrossRef]
- Kukreja, P.; Abdel-Mageed, A.B.; Mondal, D.; Liu, K.; Agrawal, K.C. Up-regulation of CXCR4 expression in PC-3 cells by stromal-derived factor-1alpha (CXCL12) increases endothelial adhesion and transendothelial migration: Role of MEK/ERK signaling pathway-dependent NF-kappaB activation. Cancer Res. 2005, 65, 9891–9898. [Google Scholar] [CrossRef]
- Engl, T.; Relja, B.; Marian, D.; Blumenberg, C.; Muller, I.; Beecken, W.D.; Jones, J.; Ringel, E.M.; Bereiter-Hahn, J.; Jonas, D.; et al. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia 2006, 8, 290–301. [Google Scholar] [CrossRef]
- Darash-Yahana, M.; Pikarsky, E.; Abramovitch, R.; Zeira, E.; Pal, B.; Karplus, R.; Beider, K.; Avniel, S.; Kasem, S.; Galum, E.; et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004, 18, 1240–1242. [Google Scholar] [CrossRef]
- Cashman, J.; Clark-Lewis, I.; Eaves, A.; Eaves, C. Stromal-derived factor 1 inhibits the cycling of very primitive human hematopoietic cells in vitro and in NOD/SCID mice. Blood 2002, 99, 792–799. [Google Scholar] [CrossRef]
- Peled, A.; Petit, I.; Kollet, O.; Magid, M.; Ponomaryov, T.; Byk, T.; Nagler, A.; Ben-Hur, H.; Many, A.; Shultz, L.; et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 1999, 283, 845–848. [Google Scholar] [CrossRef]
- Liles, W.C.; Broxmeyer, H.E.; Rodger, E.; Wood, B.; Hubel, K.; Cooper, S.; Hangoc, G.; Bridger, G.J.; Henson, G.W.; Calandra, G.; et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood 2003, 102, 2728–2730. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Burke, J.; Kolhe, R.; Hunter, M.; Isales, C.; Hamrick, M.; Fulzele, S. Stem Cell-Derived Exosomes: A Potential Alternative Therapeutic Agent in Orthopaedics. Stem Cells Int. 2016, 2016, 5802529. [Google Scholar] [CrossRef]
- Vishnubhatla, I.; Corteling, R.; Stevanato, L.; Hicks, C.; Sinden, J. The Development of Stem Cell-Derived Exosomes as a Cell-Free Regenerative Medicine. J. Circ. Biomark. 2014, 3, 2. [Google Scholar] [CrossRef]
- Shiao, S.L.; Chu, G.C.; Chung, L.W. Regulation of prostate cancer progression by the tumor microenvironment. Cancer Lett. 2016, 380, 340–348. [Google Scholar] [CrossRef]
- Théry, C.; Boussac, M.; Véron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic Analysis of Dendritic Cell-Derived Exosomes: A Secreted Subcellular Compartment Distinct from Apoptotic Vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef]
- Janowska-Wieczorek, A.; Majka, M.; Kijowski, J.; Baj-Krzyworzeka, M.; Reca, R.; Turner, A.R.; Ratajczak, J.; Emerson, S.G.; Kowalska, M.A.; Ratajczak, M.Z. Platelet-derived microparticles bind to hematopoietic stem/progenitor cells and enhance their engraftment. Blood 2001, 98, 3143–3149. [Google Scholar] [CrossRef]
- Baj-Krzyworzeka, M.; Szatanek, R.; Weglarczyk, K.; Baran, J.; Urbanowicz, B.; Branski, P.; Ratajczak, M.Z.; Zembala, M. Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol. Immunother. 2006, 55, 808–818. [Google Scholar] [CrossRef]
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M.Z. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654. [Google Scholar] [CrossRef]
- Kim, H.-S.; Choi, D.-Y.; Yun, S.J.; Choi, S.-M.; Kang, J.W.; Jung, J.W.; Hwang, D.; Kim, K.P.; Kim, D.-W. Proteomic Analysis of Microvesicles Derived from Human Mesenchymal Stem Cells. J. Proteome Res. 2012, 11, 839–849. [Google Scholar] [CrossRef]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef]
- Josson, S.; Gururajan, M.; Sung, S.Y.; Hu, P.; Shao, C.; Zhau, H.E.; Liu, C.; Lichterman, J.; Duan, P.; Li, Q.; et al. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 2014, 34, 2690. [Google Scholar] [CrossRef]
- Chowdhury, R.; Webber, J.P.; Gurney, M.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 2015, 6, 715–731. [Google Scholar] [CrossRef]
- Vlaeminck-Guillem, V. When Prostate Cancer Circulates in the Bloodstream. Diagnostics 2015, 5, 428–474. [Google Scholar] [CrossRef]
- Vlaeminck-Guillem, V. Extracellular Vesicles in Prostate Cancer Carcinogenesis, Diagnosis, and Management. Front. Oncol. 2018, 8, 222. [Google Scholar] [CrossRef]
- Bryant, R.J.; Pawlowski, T.; Catto, J.W.F.; Marsden, G.; Vessella, R.L.; Rhees, B.; Kuslich, C.; Visakorpi, T.; Hamdy, F.C. Changes in circulating microRNA levels associated with prostate cancer. Br. J. Cancer 2012, 106, 768. [Google Scholar] [CrossRef]
- Li, Z.; Ma, Y.-Y.; Wang, J.; Zeng, X.-F.; Li, R.; Kang, W.; Hao, X.-K. Exosomal microRNA-141 is upregulated in the serum of prostate cancer patients. OncoTargets Ther. 2015, 9, 139–148. [Google Scholar] [CrossRef]
- Brase, J.C.; Johannes, M.; Schlomm, T.; Falth, M.; Haese, A.; Steuber, T.; Beissbarth, T.; Kuner, R.; Sultmann, H. Circulating miRNAs are correlated with tumor progression in prostate cancer. Int. J. Cancer 2011, 128, 608–616. [Google Scholar] [CrossRef]
- Samsonov, R.; Shtam, T.; Burdakov, V.; Glotov, A.; Tsyrlina, E.; Berstein, L.; Nosov, A.; Evtushenko, V.; Filatov, M.; Malek, A. Lectin-induced agglutination method of urinary exosomes isolation followed by mi-RNA analysis: Application for prostate cancer diagnostic. Prostate 2016, 76, 68–79. [Google Scholar] [CrossRef]
- Foj, L.; Ferrer, F.; Serra, M.; Arevalo, A.; Gavagnach, M.; Gimenez, N.; Filella, X. Exosomal and Non-Exosomal Urinary miRNAs in Prostate Cancer Detection and Prognosis. Prostate 2017, 77, 573–583. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Liang, M.; Du, M.; Xia, S.; Dittmar, R.; Wang, D.; See, W.; Costello, B.A.; Quevedo, F.; et al. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur. Urol. 2015, 67, 33–41. [Google Scholar] [CrossRef]
- Corcoran, C.; Rani, S.; O’Driscoll, L. miR-34a is an intracellular and exosomal predictive biomarker for response to docetaxel with clinical relevance to prostate cancer progression. Prostate 2014, 74, 1320–1334. [Google Scholar] [CrossRef]
- Yu, Q.; Li, P.; Weng, M.; Wu, S.; Zhang, Y.; Chen, X.; Zhang, Q.; Shen, G.; Ding, X.; Fu, S. Nano-Vesicles are a Potential Tool to Monitor Therapeutic Efficacy of Carbon Ion Radiotherapy in Prostate Cancer. J. Biomed. Nanotechnol. 2018, 14, 168–178. [Google Scholar] [CrossRef]
- Itoh, T.; Ito, Y.; Ohtsuki, Y.; Ando, M.; Tsukamasa, Y.; Yamada, N.; Naoe, T.; Akao, Y. Microvesicles released from hormone-refractory prostate cancer cells facilitate mouse pre-osteoblast differentiation. J. Mol. Histol. 2012, 43, 509–515. [Google Scholar] [CrossRef]
- Karlsson, T.; Lundholm, M.; Widmark, A.; Persson, E. Tumor Cell-Derived Exosomes from the Prostate Cancer Cell Line TRAMP-C1 Impair Osteoclast Formation and Differentiation. PLoS ONE 2016, 11, e0166284. [Google Scholar] [CrossRef]
- Gonzalez, A.; Garcia de Durango, C.; Alonso, V.; Bravo, B.; Rodriguez de Gortazar, A.; Wells, A.; Forteza, J.; Vidal-Vanaclocha, F. Distinct Osteomimetic Response of Androgen-Dependent and Independent Human Prostate Cancer Cells to Mechanical Action of Fluid Flow: Prometastatic Implications. Prostate 2017, 77, 321–333. [Google Scholar] [CrossRef]
- Jadaan, D.Y.; Jadaan, M.M.; McCabe, J.P. Cellular Plasticity in Prostate Cancer Bone Metastasis. Prostate Cancer 2015, 2015, 651580. [Google Scholar] [CrossRef]
- Rucci, N.; Teti, A. Osteomimicry: How the Seed Grows in the Soil. Calcif. Tissue Int. 2018, 102, 131–140. [Google Scholar] [CrossRef]
- Shupp, A.B.; Kolb, A.D.; Mukhopadhyay, D.; Bussard, K.M. Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts. Cancers 2018, 10, 182. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Fedele, C.; Singh, A.; Zerlanko, B.J.; Iozzo, R.V.; Languino, L.R. The alphavbeta6 integrin is transferred intercellularly via exosomes. J. Biol. Chem. 2015, 290, 4545–4551. [Google Scholar] [CrossRef]
- Shimura, S.; Yang, G.; Ebara, S.; Wheeler, T.M.; Frolov, A.; Thompson, T.C. Reduced infiltration of tumor-associated macrophages in human prostate cancer: Association with cancer progression. Cancer Res. 2000, 60, 5857–5861. [Google Scholar]
- Nonomura, N.; Takayama, H.; Nakayama, M.; Nakai, Y.; Kawashima, A.; Mukai, M.; Nagahara, A.; Aozasa, K.; Tsujimura, A. Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int. 2011, 107, 1918–1922. [Google Scholar] [CrossRef]
- Lanciotti, M.; Masieri, L.; Raspollini, M.R.; Minervini, A.; Mari, A.; Comito, G.; Giannoni, E.; Carini, M.; Chiarugi, P.; Serni, S. The role of M1 and M2 macrophages in prostate cancer in relation to extracapsular tumor extension and biochemical recurrence after radical prostatectomy. Biomed. Res. Int. 2014, 2014, 486798. [Google Scholar] [CrossRef]
- Fujii, T.; Shimada, K.; Asai, O.; Tanaka, N.; Fujimoto, K.; Hirao, K.; Konishi, N. Immunohistochemical analysis of inflammatory cells in benign and precancerous lesions and carcinoma of the prostate. Pathobiology 2013, 80, 119–126. [Google Scholar] [CrossRef]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef]
- Escamilla, J.; Schokrpur, S.; Liu, C.; Priceman, S.J.; Moughon, D.; Jiang, Z.; Pouliot, F.; Magyar, C.; Sung, J.L.; Xu, J.; et al. CSF1 receptor targeting in prostate cancer reverses macrophage-mediated resistance to androgen blockade therapy. Cancer Res. 2015, 75, 950–962. [Google Scholar] [CrossRef]
- Lo, C.H.; Lynch, C.C. Multifaceted Roles for Macrophages in Prostate Cancer Skeletal Metastasis. Front. Endocrinol. 2018, 9, 247. [Google Scholar] [CrossRef]
- Mahon, K.L.; Lin, H.M.; Castillo, L.; Lee, B.Y.; Lee-Ng, M.; Chatfield, M.D.; Chiam, K.; Breit, S.N.; Brown, D.A.; Molloy, M.P.; et al. Cytokine profiling of docetaxel-resistant castration-resistant prostate cancer. Br. J. Cancer 2015, 112, 1340–1348. [Google Scholar] [CrossRef]
- Liu, G.; Lu, S.; Wang, X.; Page, S.T.; Higano, C.S.; Plymate, S.R.; Greenberg, N.M.; Sun, S.; Li, Z.; Wu, J.D. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J. Clin. Investig. 2013, 123, 4410–4422. [Google Scholar] [CrossRef]
- Gannon, P.O.; Poisson, A.O.; Delvoye, N.; Lapointe, R.; Mes-Masson, A.M.; Saad, F. Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J. Immunol. Methods 2009, 348, 9–17. [Google Scholar] [CrossRef]
- Pasero, C.; Gravis, G.; Granjeaud, S.; Guerin, M.; Thomassin-Piana, J.; Rocchi, P.; Salem, N.; Walz, J.; Moretta, A.; Olive, D. Highly effective NK cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget 2015, 6, 14360–14373. [Google Scholar] [CrossRef]
- Johansson, A.; Rudolfsson, S.; Hammarsten, P.; Halin, S.; Pietras, K.; Jones, J.; Stattin, P.; Egevad, L.; Granfors, T.; Wikstrom, P.; et al. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am. J. Pathol. 2010, 177, 1031–1041. [Google Scholar] [CrossRef]
- Fleischmann, A.; Schlomm, T.; Kollermann, J.; Sekulic, N.; Huland, H.; Mirlacher, M.; Sauter, G.; Simon, R.; Erbersdobler, A. Immunological microenvironment in prostate cancer: High mast cell densities are associated with favorable tumor characteristics and good prognosis. Prostate 2009, 69, 976–981. [Google Scholar] [CrossRef]
- Pittoni, P.; Tripodo, C.; Piconese, S.; Mauri, G.; Parenza, M.; Rigoni, A.; Sangaletti, S.; Colombo, M.P. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res. 2011, 71, 5987–5997. [Google Scholar] [CrossRef]
- Templeton, A.J.; Pezaro, C.; Omlin, A.; McNamara, M.G.; Leibowitz-Amit, R.; Vera-Badillo, F.E.; Attard, G.; de Bono, J.S.; Tannock, I.F.; Amir, E. Simple prognostic score for metastatic castration-resistant prostate cancer with incorporation of neutrophil-to-lymphocyte ratio. Cancer 2014, 120, 3346–3352. [Google Scholar] [CrossRef]
- Ong, C.W.; Pabisiak, P.J.; Brilha, S.; Singh, P.; Roncaroli, F.; Elkington, P.T.; Friedland, J.S. Complex regulation of neutrophil-derived MMP-9 secretion in central nervous system tuberculosis. J. Neuroinflamm. 2017, 14, 31. [Google Scholar] [CrossRef]
- Fang, L.Y.; Izumi, K.; Lai, K.P.; Liang, L.; Li, L.; Miyamoto, H.; Lin, W.J.; Chang, C. Infiltrating macrophages promote prostate tumorigenesis via modulating androgen receptor-mediated CCL4-STAT3 signaling. Cancer Res. 2013, 73, 5633–5646. [Google Scholar] [CrossRef]
- Pittoni, P.; Colombo, M.P. The dark side of mast cell-targeted therapy in prostate cancer. Cancer Res. 2012, 72, 831–835. [Google Scholar] [CrossRef]
- Taverna, G.; Giusti, G.; Seveso, M.; Hurle, R.; Colombo, P.; Stifter, S.; Grizzi, F. Mast cells as a potential prognostic marker in prostate cancer. Dis. Markers 2013, 35, 711–720. [Google Scholar] [CrossRef]
- Okada, K.; Kojima, M.; Naya, Y.; Kamoi, K.; Yokoyama, K.; Takamatsu, T.; Miki, T. Correlation of histological inflammation in needle biopsy specimens with serum prostate- specific antigen levels in men with negative biopsy for prostate cancer. Urology 2000, 55, 892–898. [Google Scholar] [CrossRef]
- Kwek, S.S.; Lewis, J.; Zhang, L.; Weinberg, V.; Greaney, S.K.; Harzstark, A.L.; Lin, A.M.; Ryan, C.J.; Small, E.J.; Fong, L. Preexisting Levels of CD4 T Cells Expressing PD-1 Are Related to Overall Survival in Prostate Cancer Patients Treated with Ipilimumab. Cancer Immunol. Res. 2015, 3, 1008–1016. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- McArdle, P.A.; Canna, K.; McMillan, D.C.; McNicol, A.M.; Campbell, R.; Underwood, M.A. The relationship between T-lymphocyte subset infiltration and survival in patients with prostate cancer. Br. J. Cancer 2004, 91, 541–543. [Google Scholar] [CrossRef]
- Ness, N.; Andersen, S.; Valkov, A.; Nordby, Y.; Donnem, T.; Al-Saad, S.; Busund, L.T.; Bremnes, R.M.; Richardsen, E. Infiltration of CD8+ lymphocytes is an independent prognostic factor of biochemical failure-free survival in prostate cancer. Prostate 2014, 74, 1452–1461. [Google Scholar] [CrossRef]
- Liu, Y.; Saeter, T.; Vlatkovic, L.; Servoll, E.; Waaler, G.; Axcrona, U.; Giercksky, K.E.; Nesland, J.M.; Suo, Z.H.; Axcrona, K. Dendritic and lymphocytic cell infiltration in prostate carcinoma. Histol. Histopathol. 2013, 28, 1621–1628. [Google Scholar] [CrossRef]
- Davidsson, S.; Ohlson, A.L.; Andersson, S.O.; Fall, K.; Meisner, A.; Fiorentino, M.; Andren, O.; Rider, J.R. CD4 helper T cells, CD8 cytotoxic T cells, and FOXP3(+) regulatory T cells with respect to lethal prostate cancer. Mod. Pathol. 2013, 26, 448–455. [Google Scholar] [CrossRef]
- Ebelt, K.; Babaryka, G.; Frankenberger, B.; Stief, C.G.; Eisenmenger, W.; Kirchner, T.; Schendel, D.J.; Noessner, E. Prostate cancer lesions are surrounded by FOXP3+, PD-1+ and B7-H1+ lymphocyte clusters. Eur. J. Cancer 2009, 45, 1664–1672. [Google Scholar] [CrossRef]
- Miller, A.M.; Lundberg, K.; Ozenci, V.; Banham, A.H.; Hellstrom, M.; Egevad, L.; Pisa, P. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J. Immunol. 2006, 177, 7398–7405. [Google Scholar] [CrossRef]
- Idorn, M.; Kollgaard, T.; Kongsted, P.; Sengelov, L.; Thor Straten, P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol. Immunother. 2014, 63, 1177–1187. [Google Scholar] [CrossRef]
- Kumar, P.; Saini, S.; Prabhakar, B.S. Cancer immunotherapy with check point inhibitor can cause autoimmune adverse events due to loss of Treg homeostasis. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Affara, N.I.; Ruffell, B.; Medler, T.R.; Gunderson, A.J.; Johansson, M.; Bornstein, S.; Bergsland, E.; Steinhoff, M.; Li, Y.; Gong, Q.; et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell 2014, 25, 809–821. [Google Scholar] [CrossRef]
- Woo, J.R.; Liss, M.A.; Muldong, M.T.; Palazzi, K.; Strasner, A.; Ammirante, M.; Varki, N.; Shabaik, A.; Howell, S.; Kane, C.J.; et al. Tumor infiltrating B-cells are increased in prostate cancer tissue. J. Transl. Med. 2014, 12, 30. [Google Scholar] [CrossRef]
- Strasner, A.; Karin, M. Immune Infiltration and Prostate Cancer. Front. Oncol. 2015, 5, 128. [Google Scholar] [CrossRef]
- Ammirante, M.; Kuraishy, A.I.; Shalapour, S.; Strasner, A.; Ramirez-Sanchez, C.; Zhang, W.; Shabaik, A.; Karin, M. An IKKalpha-E2F1-BMI1 cascade activated by infiltrating B cells controls prostate regeneration and tumor recurrence. Genes Dev. 2013, 27, 1435–1440. [Google Scholar] [CrossRef]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E.; et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef]
- The spread of tumours in the human body. By Rupert A. Willis. M.D., B.S., D.Sc. (Melbourne), Pathologist to the Alfred Hospital and to the Austin Hospital for Chronic Diseases, Melbourne. Demy 8vo. Pp. 540 + x, with 103 illustrations. 1934 London: J. & A. Churchill. 25s. net. BJS 1934, 22, 196. [CrossRef]
- Schlimok, G.; Funke, I.; Holzmann, B.; Gottlinger, G.; Schmidt, G.; Hauser, H.; Swierkot, S.; Warnecke, H.H.; Schneider, B.; Koprowski, H.; et al. Micrometastatic cancer cells in bone marrow: In vitro detection with anti-cytokeratin and in vivo labeling with anti-17-1A monoclonal antibodies. Proc. Natl. Acad. Sci. USA 1987, 84, 8672–8676. [Google Scholar] [CrossRef]
- Cote, R.J.; Rosen, P.P.; Hakes, T.B.; Sedira, M.; Bazinet, M.; Kinne, D.W.; Old, L.J.; Osborne, M.P. Monoclonal antibodies detect occult breast carcinoma metastases in the bone marrow of patients with early stage disease. Am. J. Surg. Pathol. 1988, 12, 333–340. [Google Scholar] [CrossRef]
- Pantel, K.; Izbicki, J.; Passlick, B.; Angstwurm, M.; Haussinger, K.; Thetter, O.; Riethmuller, G. Frequency and prognostic significance of isolated tumour cells in bone marrow of patients with non-small-cell lung cancer without overt metastases. Lancet 1996, 347, 649–653. [Google Scholar] [CrossRef]
- Husemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmuller, G.; et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef]
- Cote, R.J.; Rosen, P.P.; Lesser, M.L.; Old, L.J.; Osborne, M.P. Prediction of early relapse in patients with operable breast cancer by detection of occult bone marrow micrometastases. J. Clin. Oncol. 1991, 9, 1749–1756. [Google Scholar] [CrossRef]
- Mansi, J.L.; Easton, D.; Berger, U.; Gazet, J.C.; Ford, H.T.; Dearnaley, D.; Coombes, R.C. Bone marrow micrometastases in primary breast cancer: Prognostic significance after 6 years’ follow-up. Eur. J. Cancer 1991, 27, 1552–1555. [Google Scholar] [CrossRef]
- Pantel, K.; Aignherr, C.; Kollermann, J.; Caprano, J.; Riethmuller, G.; Kollermann, M.W. Immunocytochemical detection of isolated tumour cells in bone marrow of patients with untreated stage C prostatic cancer. Eur. J. Cancer 1995, 31A, 1627–1632. [Google Scholar] [CrossRef]
- Thorban, S.; Roder, J.D.; Nekarda, H.; Funk, A.; Siewert, J.R.; Pantel, K. Immunocytochemical detection of disseminated tumor cells in the bone marrow of patients with esophageal carcinoma. J. Natl. Cancer Inst. 1996, 88, 1222–1227. [Google Scholar] [CrossRef]
- Schlimok, G.; Funke, I.; Pantel, K.; Strobel, F.; Lindemann, F.; Witte, J.; Riethmuller, G. Micrometastatic tumour cells in bone marrow of patients with gastric cancer: Methodological aspects of detection and prognostic significance. Eur. J. Cancer 1991, 27, 1461–1465. [Google Scholar] [CrossRef]
- Weckermann, D.; Muller, P.; Wawroschek, F.; Harzmann, R.; Riethmuller, G.; Schlimok, G. Disseminated cytokeratin positive tumor cells in the bone marrow of patients with prostate cancer: Detection and prognostic value. J. Urol. 2001, 166, 699–703. [Google Scholar] [CrossRef]
- Goss, P.E.; Chambers, A.F. Does tumour dormancy offer a therapeutic target? Nat. Rev. Cancer 2010, 10, 871–877. [Google Scholar] [CrossRef]
- Meng, S.; Tripathy, D.; Frenkel, E.P.; Shete, S.; Naftalis, E.Z.; Huth, J.F.; Beitsch, P.D.; Leitch, M.; Hoover, S.; Euhus, D.; et al. Circulating tumor cells in patients with breast cancer dormancy. Clin. Cancer Res. 2004, 10, 8152–8162. [Google Scholar] [CrossRef]
- Uhr, J.W.; Pantel, K. Controversies in clinical cancer dormancy. Proc. Natl. Acad. Sci. USA 2011, 108, 12396–12400. [Google Scholar] [CrossRef]
- Kang, Y.; Pantel, K. Tumor cell dissemination: Emerging biological insights from animal models and cancer patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef]
- Naumov, G.N.; MacDonald, I.C.; Weinmeister, P.M.; Kerkvliet, N.; Nadkarni, K.V.; Wilson, S.M.; Morris, V.L.; Groom, A.C.; Chambers, A.F. Persistence of solitary mammary carcinoma cells in a secondary site: A possible contributor to dormancy. Cancer Res. 2002, 62, 2162–2168. [Google Scholar]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell. Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef]
- Jung, Y.; Shiozawa, Y.; Wang, J.; McGregor, N.; Dai, J.; Park, S.I.; Berry, J.E.; Havens, A.M.; Joseph, J.; Kim, J.K.; et al. Prevalence of prostate cancer metastases after intravenous inoculation provides clues into the molecular basis of dormancy in the bone marrow microenvironment. Neoplasia 2012, 14, 429–439. [Google Scholar] [CrossRef]
- Taichman, R.S.; Patel, L.R.; Bedenis, R.; Wang, J.; Weidner, S.; Schumann, T.; Yumoto, K.; Berry, J.E.; Shiozawa, Y.; Pienta, K.J. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS ONE 2013, 8, e61873. [Google Scholar] [CrossRef]
- Mishra, A.; Wang, J.; Shiozawa, Y.; McGee, S.; Kim, J.; Jung, Y.; Joseph, J.; Berry, J.E.; Havens, A.; Pienta, K.J.; et al. Hypoxia stabilizes GAS6/Axl signaling in metastatic prostate cancer. Mol. Cancer Res. 2012, 10, 703–712. [Google Scholar] [CrossRef]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef]
- Guise, T.A.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef]
- Kollet, O.; Dar, A.; Shivtiel, S.; Kalinkovich, A.; Lapid, K.; Sztainberg, Y.; Tesio, M.; Samstein, R.M.; Goichberg, P.; Spiegel, A.; et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat. Med. 2006, 12, 657–664. [Google Scholar] [CrossRef]
- Michaelson, M.D.; Marujo, R.M.; Smith, M.R. Contribution of androgen deprivation therapy to elevated osteoclast activity in men with metastatic prostate cancer. Clin. Cancer Res. 2004, 10, 2705–2708. [Google Scholar] [CrossRef][Green Version]
- Smith, M.R.; McGovern, F.J.; Zietman, A.L.; Fallon, M.A.; Hayden, D.L.; Schoenfeld, D.A.; Kantoff, P.W.; Finkelstein, J.S. Pamidronate to prevent bone loss during androgen-deprivation therapy for prostate cancer. N. Engl. J. Med. 2001, 345, 948–955. [Google Scholar] [CrossRef]
- Smith, M.R.; Eastham, J.; Gleason, D.M.; Shasha, D.; Tchekmedyian, S.; Zinner, N. Randomized controlled trial of zoledronic acid to prevent bone loss in men receiving androgen deprivation therapy for nonmetastatic prostate cancer. J. Urol. 2003, 169, 2008–2012. [Google Scholar] [CrossRef]
- Greenspan, S.L.; Nelson, J.B.; Trump, D.L.; Resnick, N.M. Effect of once-weekly oral alendronate on bone loss in men receiving androgen deprivation therapy for prostate cancer: A randomized trial. Ann. Intern. Med. 2007, 146, 416–424. [Google Scholar] [CrossRef]
- Klotz, L.H.; McNeill, I.Y.; Kebabdjian, M.; Zhang, L.; Chin, J.L.; Canadian Urology Research, C. A phase 3, double-blind, randomised, parallel-group, placebo-controlled study of oral weekly alendronate for the prevention of androgen deprivation bone loss in nonmetastatic prostate cancer: The Cancer and Osteoporosis Research with Alendronate and Leuprolide (CORAL) study. Eur. Urol. 2013, 63, 927–935. [Google Scholar] [CrossRef]
- Smith, M.R.; Egerdie, B.; Hernandez Toriz, N.; Feldman, R.; Tammela, T.L.; Saad, F.; Heracek, J.; Szwedowski, M.; Ke, C.; Kupic, A.; et al. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N. Engl. J. Med. 2009, 361, 745–755. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Egerdie, B.; Sieber, P.; Tammela, T.; Leder, B.Z.; Ke, C.; Goessl, C. Denosumab and changes in bone turnover markers during androgen deprivation therapy for prostate cancer. J. Bone Miner. Res. 2011, 26, 2827–2833. [Google Scholar] [CrossRef]
- Ottewell, P.D.; Wang, N.; Meek, J.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. Castration-induced bone loss triggers growth of disseminated prostate cancer cells in bone. Endocr. Relat. Cancer 2014, 21, 769–781. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Coleman, R.; Shore, N.; Fizazi, K.; Tombal, B.; Miller, K.; Sieber, P.; Karsh, L.; Damiao, R.; et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial. Lancet 2012, 379, 39–46. [Google Scholar] [CrossRef]
- Nozawa, M.; Inagaki, T.; Nagao, K.; Nishioka, T.; Komura, T.; Esa, A.; Kitagawa, M.; Imanishi, M.; Uekado, Y.; Ogawa, T.; et al. Phase II trial of zoledronic acid combined with androgen-deprivation therapy for treatment-naive prostate cancer with bone metastasis. Int. J. Clin. Oncol. 2014, 19, 693–701. [Google Scholar] [CrossRef]
- Okegawa, T.; Higaki, M.; Matsumoto, T.; Kase, H.; Murata, A.; Noda, K.; Noda, H.; Asaoka, H.; Oshi, M.; Tomoishi, J.; et al. Zoledronic acid improves clinical outcomes in patients with bone metastatic hormone-naive prostate cancer in a multicenter clinical trial. Anticancer Res. 2014, 34, 4415–4420. [Google Scholar]
- Smith, M.R.; Halabi, S.; Ryan, C.J.; Hussain, A.; Vogelzang, N.; Stadler, W.; Hauke, R.J.; Monk, J.P.; Saylor, P.; Bhoopalam, N.; et al. Randomized controlled trial of early zoledronic acid in men with castration-sensitive prostate cancer and bone metastases: Results of CALGB 90202 (alliance). J. Clin. Oncol. 2014, 32, 1143–1150. [Google Scholar] [CrossRef]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362. [Google Scholar] [CrossRef]
- Makova, K.D.; Hardison, R.C. The effects of chromatin organization on variation in mutation rates in the genome. Nat. Rev. Genet. 2015, 16, 213–223. [Google Scholar] [CrossRef]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef]
- Taipale, J. The chromatin of cancer. Science 2018, 362, 401–402. [Google Scholar] [CrossRef]
- Urbanucci, A.; Barfeld, S.J.; Kytola, V.; Itkonen, H.M.; Coleman, I.M.; Vodak, D.; Sjoblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen Receptor Deregulation Drives Bromodomain-Mediated Chromatin Alterations in Prostate Cancer. Cell Rep. 2017, 19, 2045–2059. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.A.; et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 2015, 47, 1346–1351. [Google Scholar] [CrossRef]
- Stelloo, S.; Nevedomskaya, E.; van der Poel, H.G.; de Jong, J.; van Leenders, G.J.; Jenster, G.; Wessels, L.F.; Bergman, A.M.; Zwart, W. Androgen receptor profiling predicts prostate cancer outcome. EMBO Mol. Med. 2015, 7, 1450–1464. [Google Scholar] [CrossRef]
- Cirillo, L.A.; Lin, F.R.; Cuesta, I.; Friedman, D.; Jarnik, M.; Zaret, K.S. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 2002, 9, 279–289. [Google Scholar] [CrossRef]
- Shim, E.Y.; Woodcock, C.; Zaret, K.S. Nucleosome positioning by the winged helix transcription factor HNF3. Genes Dev. 1998, 12, 5–10. [Google Scholar] [CrossRef]
- Sekiya, T.; Muthurajan, U.M.; Luger, K.; Tulin, A.V.; Zaret, K.S. Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 2009, 23, 804–809. [Google Scholar] [CrossRef]
- He, H.H.; Meyer, C.A.; Shin, H.; Bailey, S.T.; Wei, G.; Wang, Q.; Zhang, Y.; Xu, K.; Ni, M.; Lupien, M.; et al. Nucleosome dynamics define transcriptional enhancers. Nat. Genet. 2010, 42, 343–347. [Google Scholar] [CrossRef]
- Gerhardt, J.; Montani, M.; Wild, P.; Beer, M.; Huber, F.; Hermanns, T.; Muntener, M.; Kristiansen, G. FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am. J. Pathol. 2012, 180, 848–861. [Google Scholar] [CrossRef]
- Gao, N.; Zhang, J.; Rao, M.A.; Case, T.C.; Mirosevich, J.; Wang, Y.; Jin, R.; Gupta, A.; Rennie, P.S.; Matusik, R.J. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 2003, 17, 1484–1507. [Google Scholar] [CrossRef]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888. [Google Scholar] [CrossRef]
- Aparicio, A.; Logothetis, C.J.; Maity, S.N. Understanding the lethal variant of prostate cancer: Power of examining extremes. Cancer Discov. 2011, 1, 466–468. [Google Scholar] [CrossRef]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive variants of castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 2846–2850. [Google Scholar] [CrossRef]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- McNair, C.; Xu, K.; Mandigo, A.C.; Benelli, M.; Leiby, B.; Rodrigues, D.; Lindberg, J.; Gronberg, H.; Crespo, M.; De Laere, B.; et al. Differential impact of RB status on E2F1 reprogramming in human cancer. J. Clin. Investig. 2018, 128, 341–358. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef]
- Clermont, P.L.; Lin, D.; Crea, F.; Wu, R.; Xue, H.; Wang, Y.; Thu, K.L.; Lam, W.L.; Collins, C.C.; Wang, Y.; et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin. Epigenet. 2015, 7, 40. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tharp, D.; Nandana, S. How Prostate Cancer Cells Use Strategy Instead of Brute Force to Achieve Metastasis. Cancers 2019, 11, 1928. https://doi.org/10.3390/cancers11121928
Tharp D, Nandana S. How Prostate Cancer Cells Use Strategy Instead of Brute Force to Achieve Metastasis. Cancers. 2019; 11(12):1928. https://doi.org/10.3390/cancers11121928
Chicago/Turabian StyleTharp, Darron, and Srinivas Nandana. 2019. "How Prostate Cancer Cells Use Strategy Instead of Brute Force to Achieve Metastasis" Cancers 11, no. 12: 1928. https://doi.org/10.3390/cancers11121928
APA StyleTharp, D., & Nandana, S. (2019). How Prostate Cancer Cells Use Strategy Instead of Brute Force to Achieve Metastasis. Cancers, 11(12), 1928. https://doi.org/10.3390/cancers11121928