PI3K/AKT/β-Catenin Signaling Regulates Vestigial-Like 1 Which Predicts Poor Prognosis and Enhances Malignant Phenotype in Gastric Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
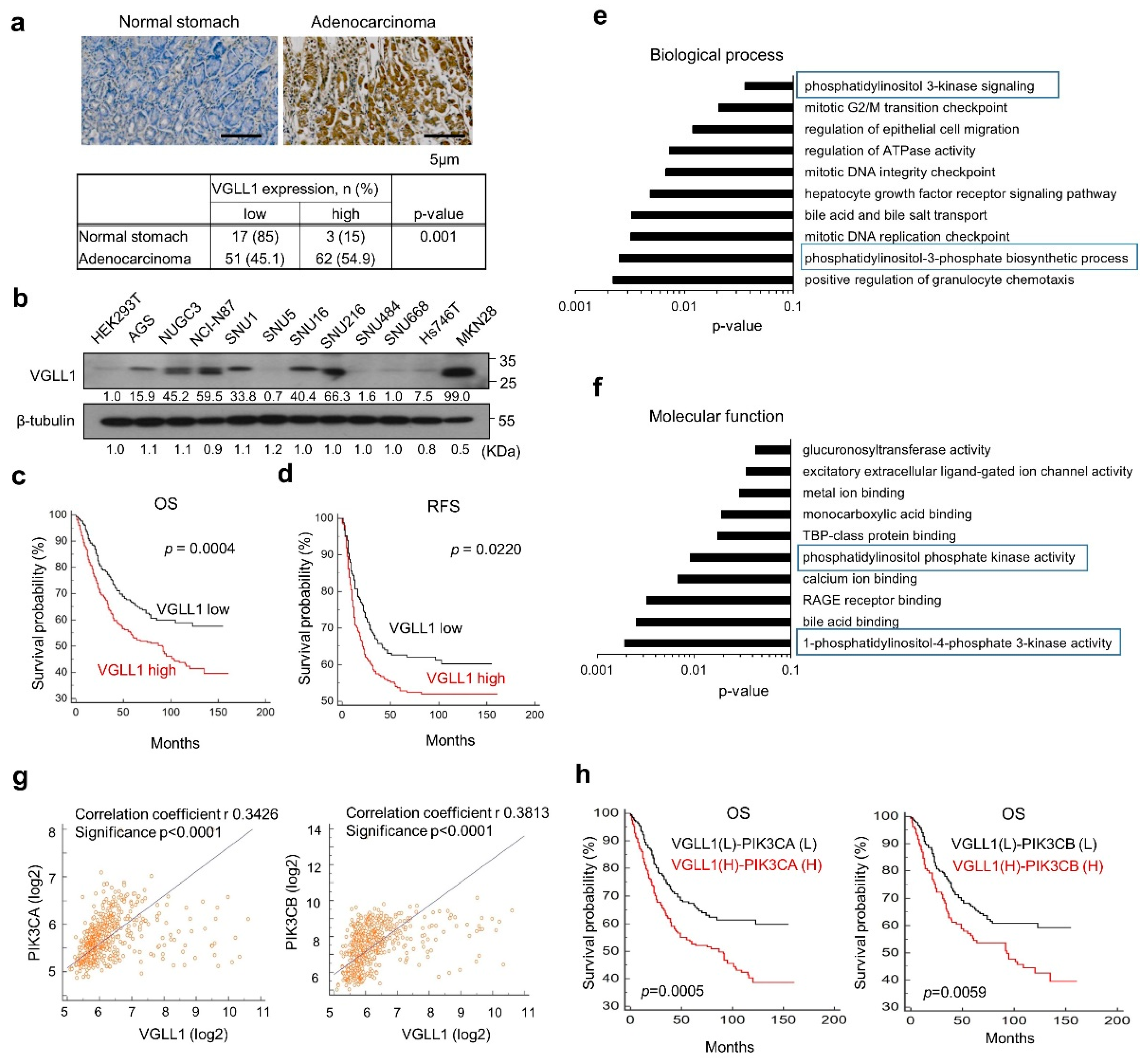
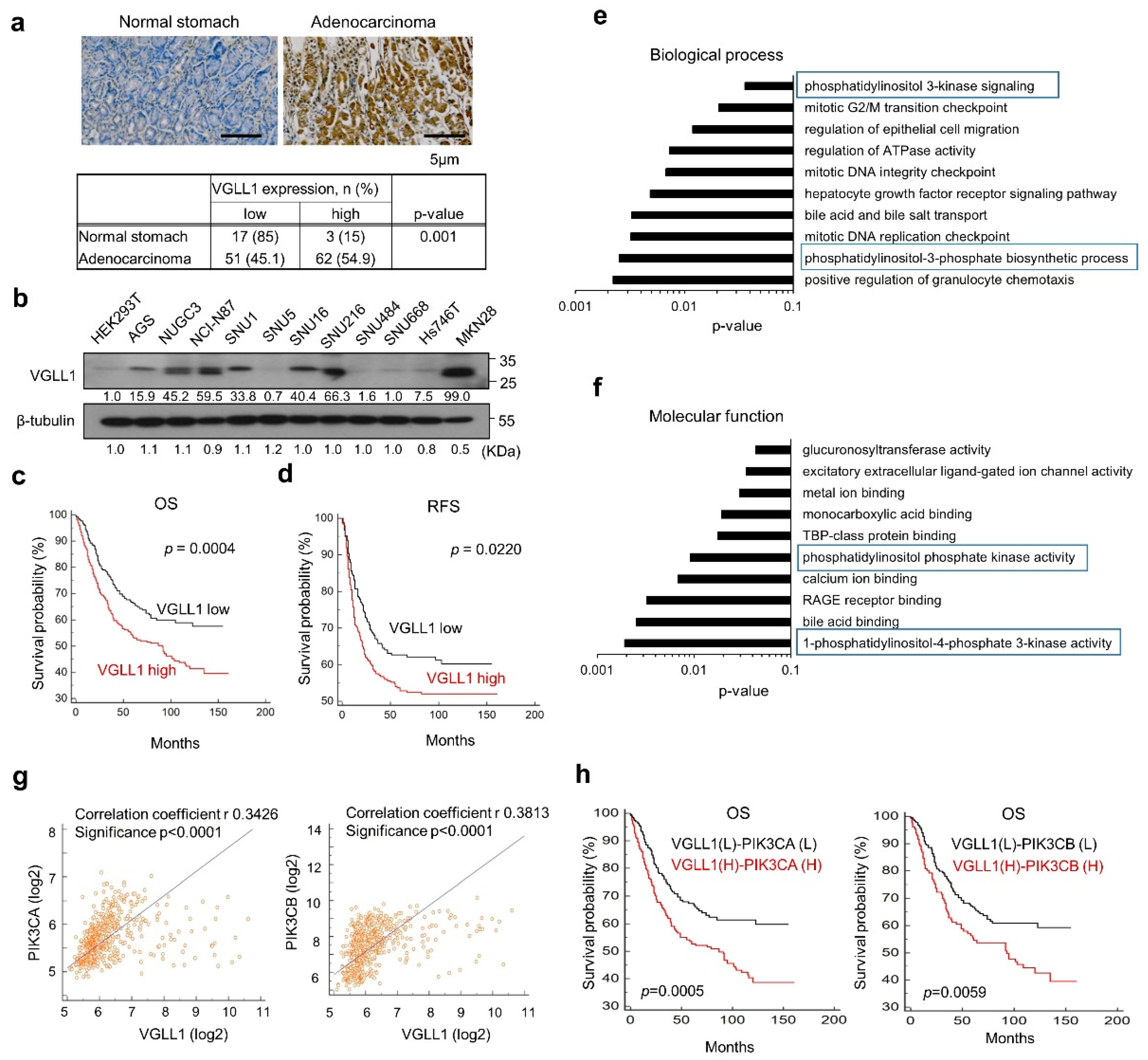
2.1. VGLL1 Is a Novel Prognostic Biomarker Correlated with PIK3CA in Gastric Cancer
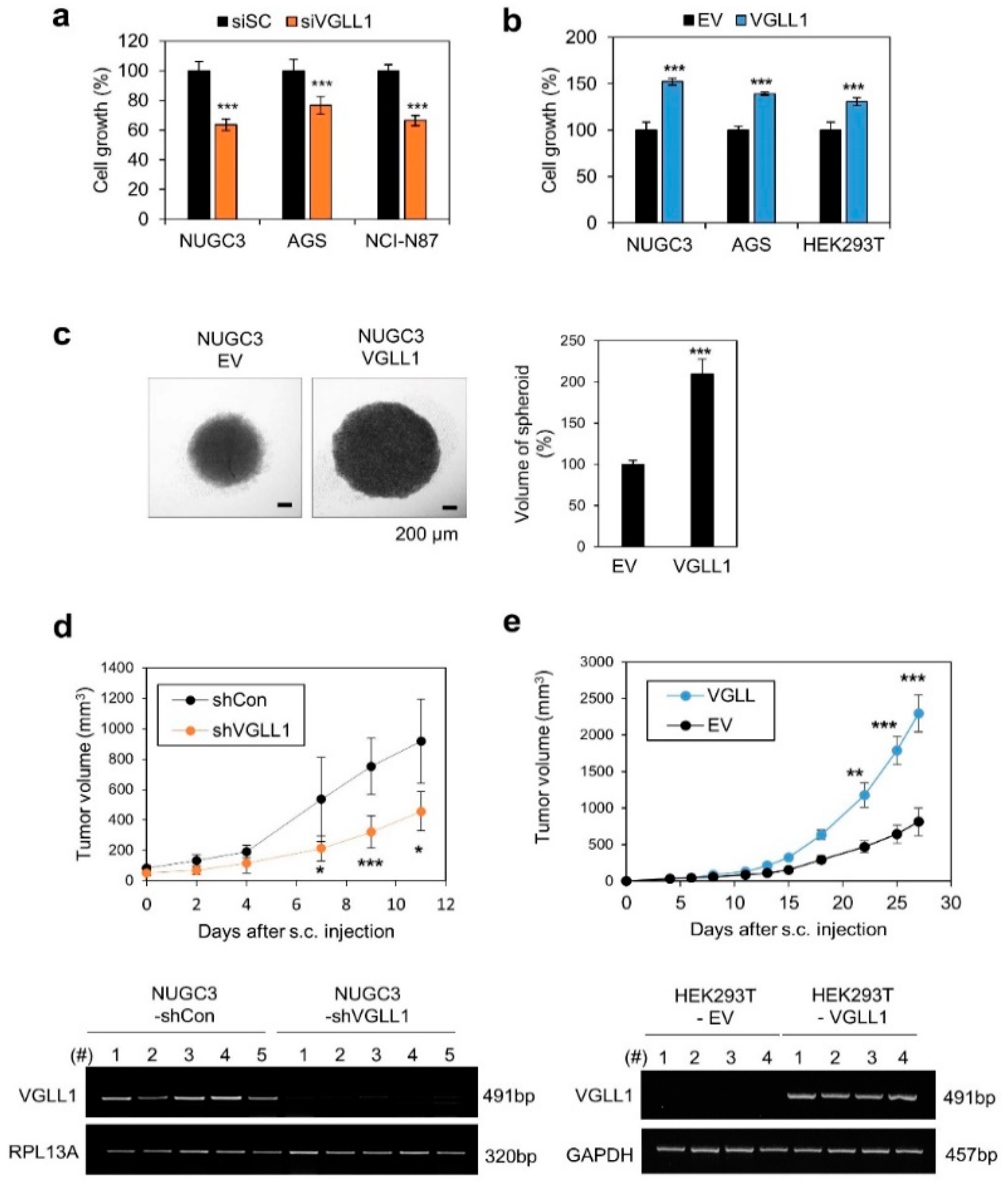
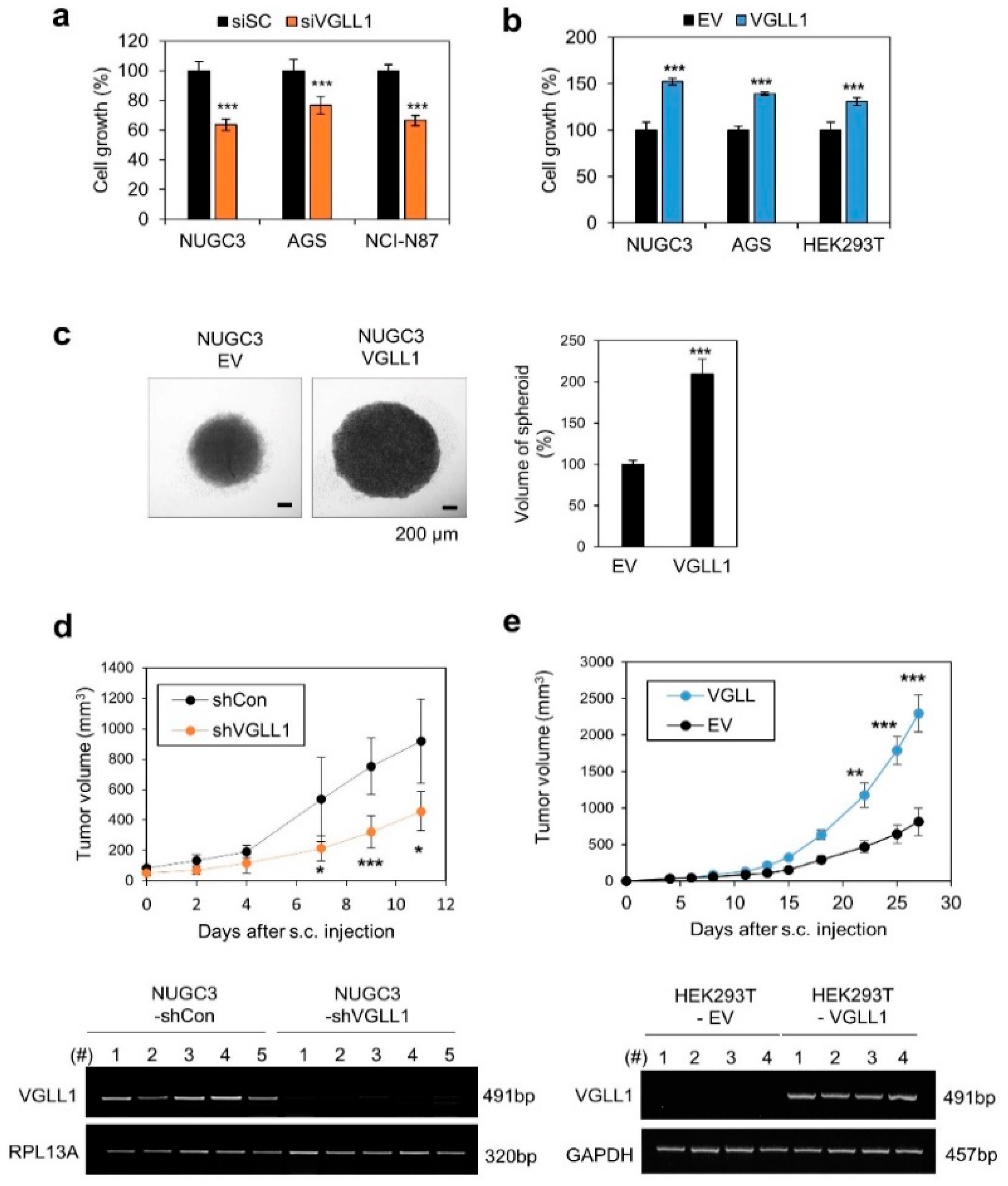
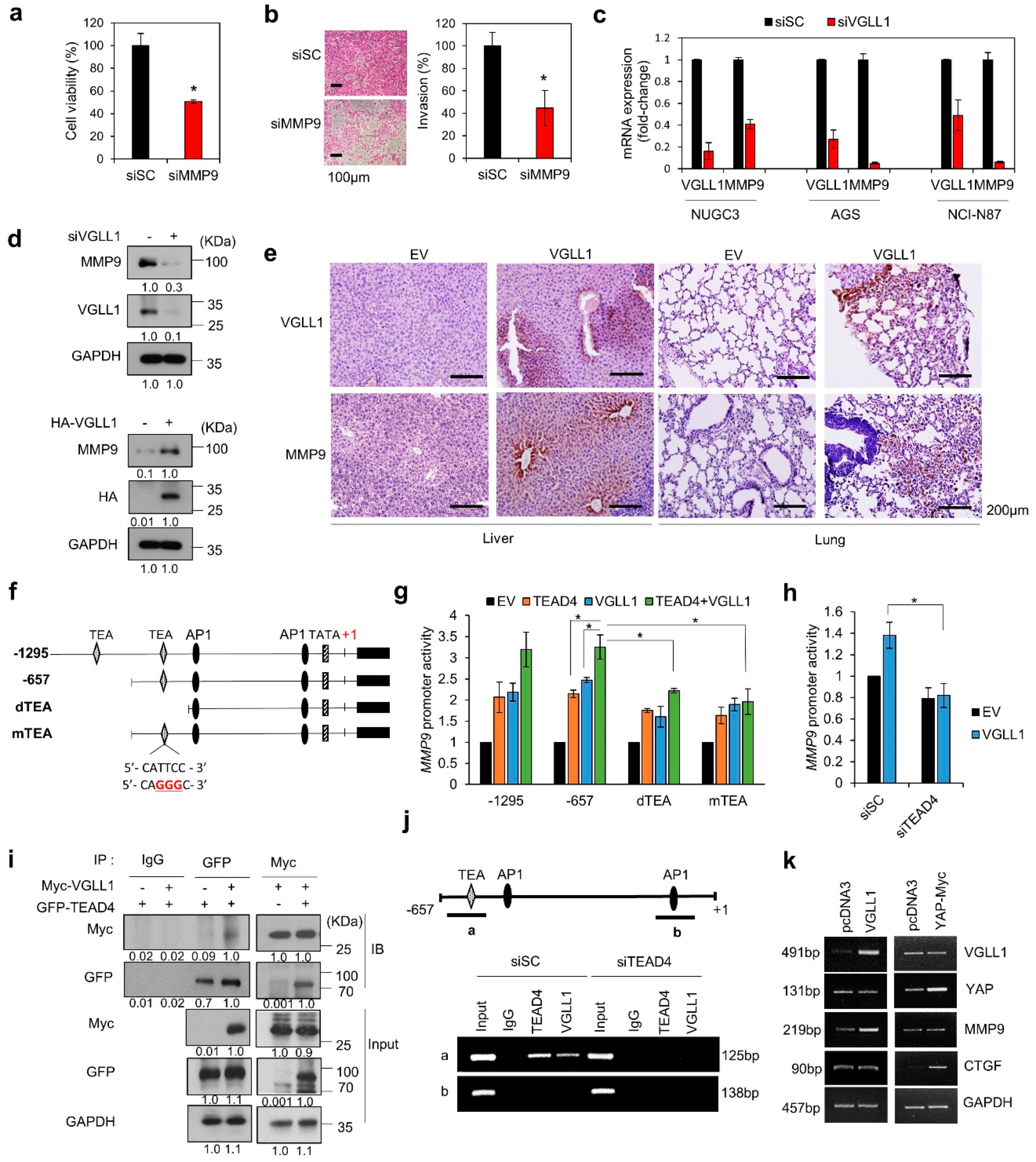
2.2. VGLL1 Regulates the Proliferation of Gastric Cancer Cells
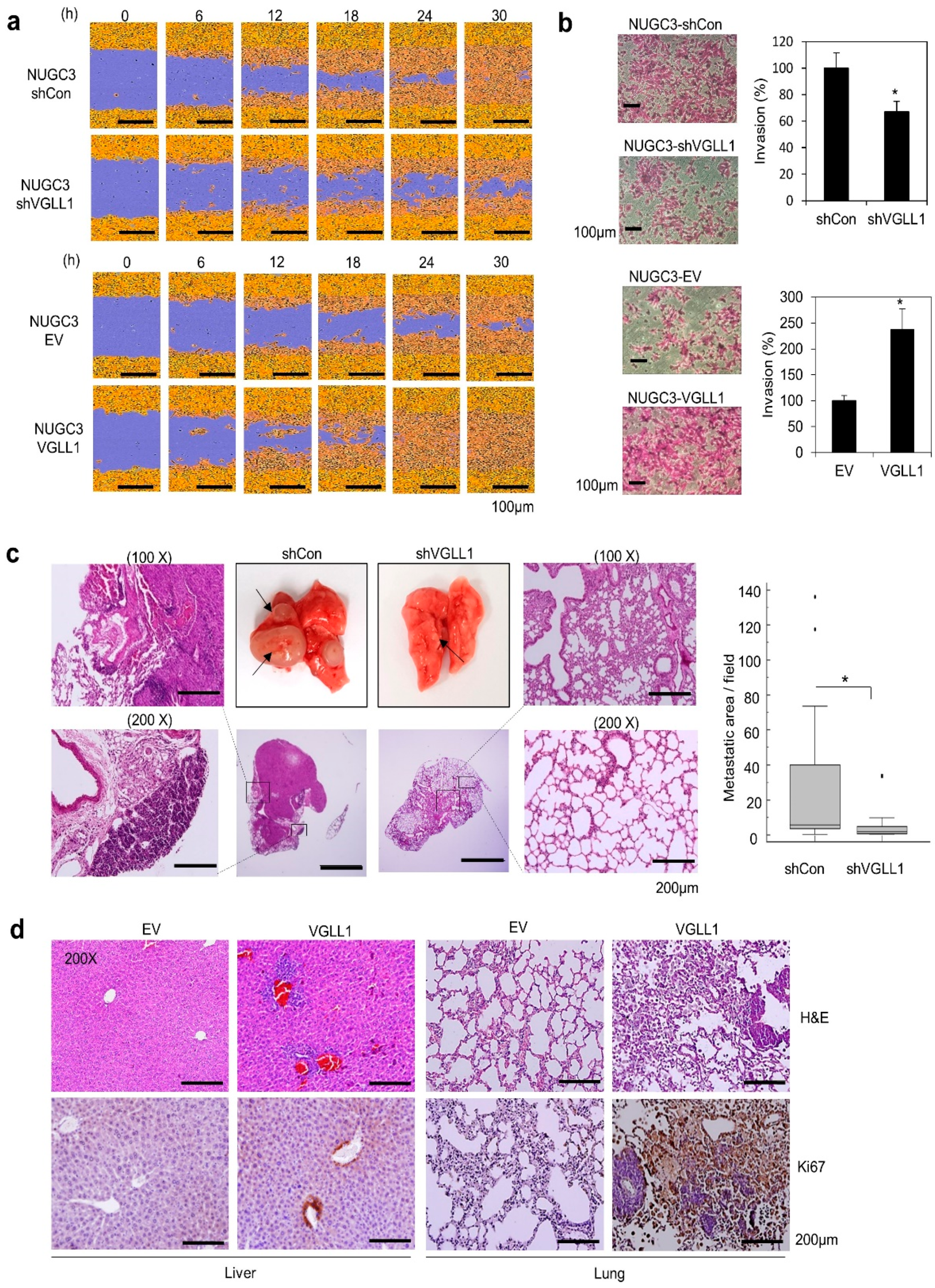
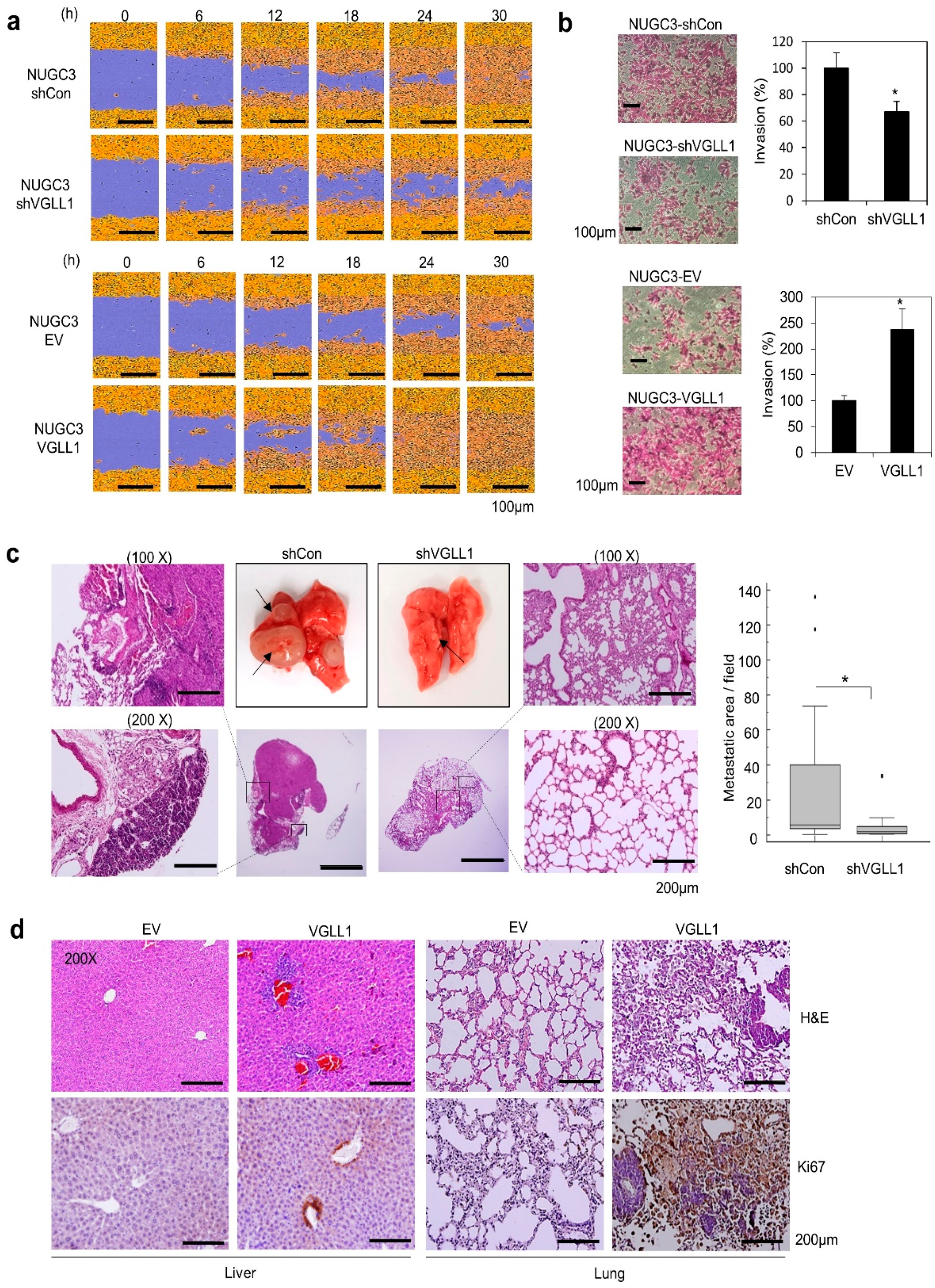
2.3. VGLL1 Regulates Metastasis of Gastric Cancer Cells in in Vitro and in Vivo Mouse Models
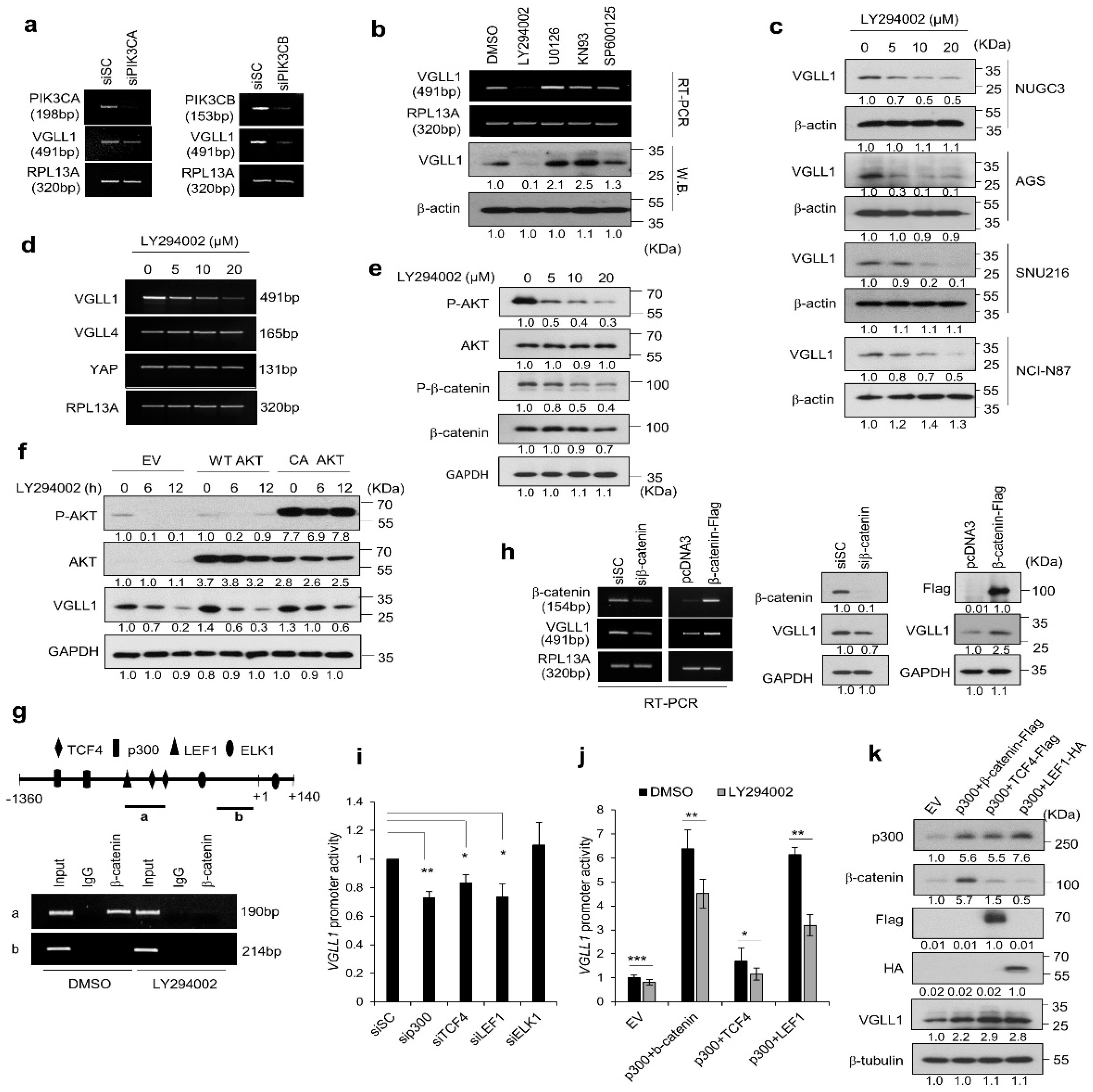
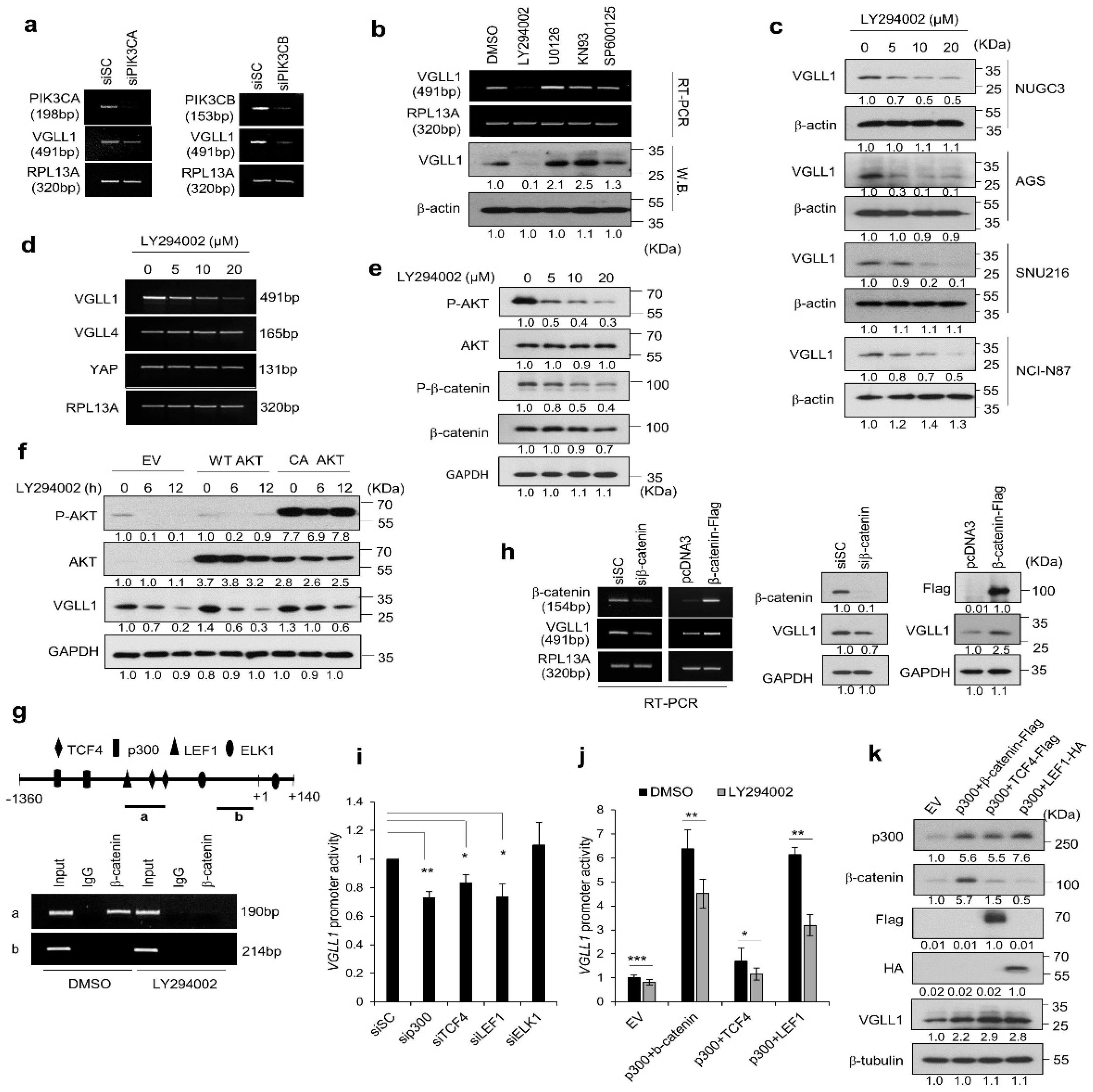
2.4. PI3K/AKT/β-Catenin Signaling Participates in the Regulation of VGLL1 Expression
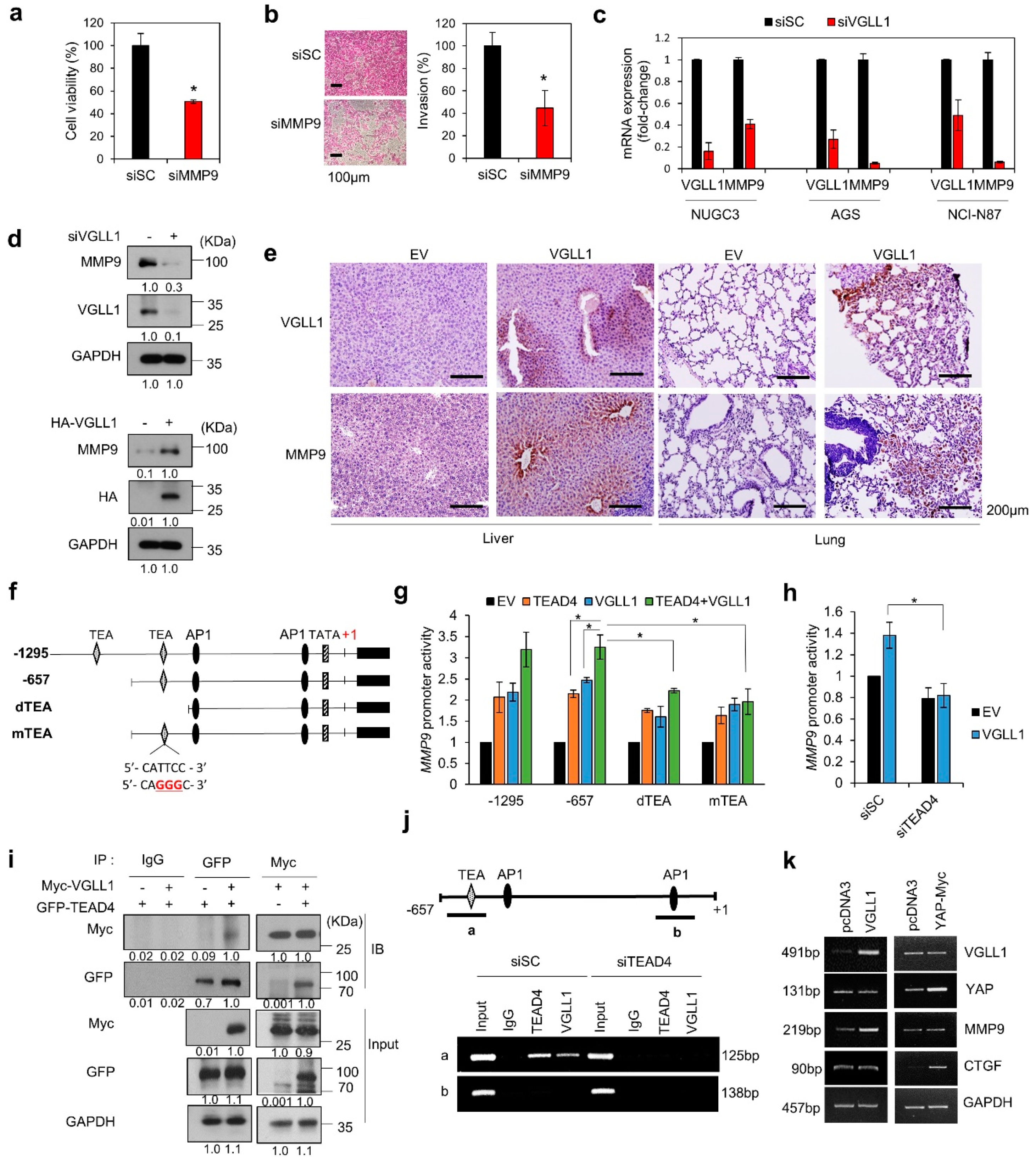
2.5. VGLL1, a TEAD4 Cofactor, Regulates MMP9 Transcription
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Patients
4.4. Microarray Experiments and Data Processing
4.5. Statistical Analysis of Microarray
4.6. Plasmids Construction
4.7. Live cell Assay for Cell Proliferation and Migration
4.8. Spheroid Formation
4.9. Mice Experiments
4.10. Western Blot Analysis
4.11. Reverse Transcriptase Polymerase Chain Reaction and Quantitative Real-Time PCR
4.12. Gene Knockdown Using siRNA
4.13. Lentivirus Infection
4.14. Immunohistochemistry (IHC)
4.15. Invasion Assay
4.16. Luciferase Assay
4.17. Chromatin Immunoprecipitation (ChIP) Assays
4.18. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheong, J.H.; Yang, H.K.; Kim, H.; Kim, W.H.; Kim, Y.W.; Kook, M.C.; Park, Y.K.; Kim, H.H.; Lee, H.S.; Lee, K.H.; et al. Predictive test for chemotherapy response in resectable gastric cancer: A multi-cohort, retrospective analysis. Lancet Oncol. 2018, 19, 629–638. [Google Scholar] [CrossRef]
- Kelly, C.M.; Janjigian, Y.Y. The genomics and therapeutics of HER2-positive gastric cancer-from trastuzumab and beyond. J. Gastrointest. Oncol. 2016, 7, 750–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabsch, H.; Sivakumar, S.; Gray, S.; Gabbert, H.E.; Muller, W. HER2 expression in gastric cancer: Rare, heterogeneous and of no prognostic value-conclusions from 924 cases of two independent series. Cell Oncol. 2010, 32, 57–65. [Google Scholar]
- Boku, N. HER2-positive gastric cancer. Gastric Cancer 2014, 17, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apicella, M.; Corso, S.; Giordano, S. Targeted therapies for gastric cancer: Failures and hopes from clinical trials. Oncotarget 2017, 8, 57654–57669. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Li, J. Current Molecular Targeted Therapy in Advanced Gastric Cancer: A Comprehensive Review of Therapeutic Mechanism, Clinical Trials, and Practical Application. Gastroenterol. Res. Pract. 2016, 2016, 4105615. [Google Scholar] [CrossRef] [Green Version]
- Baniak, N.; Senger, J.L.; Ahmed, S.; Kanthan, S.C.; Kanthan, R. Gastric biomarkers: A global review. World J. Surg. Oncol. 2016, 14, 212. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.; Yau, T. Molecular targeted therapies in advanced gastric cancer: Does tumor histology matter? Therap. Adv. Gastroenterol. 2013, 6, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Yeoh, K.G. Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology 2015, 149, 1153–1162.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacome, A.A.; Coutinho, A.K.; Lima, E.M.; Andrade, A.C.; Dos Santos, J.S. Personalized medicine in gastric cancer: Where are we and where are we going? World J. Gastroenterol. 2016, 22, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Diaz, L.A. Jr.; Schmidt-Kittler, O.; Cummins, J.M.; Delong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, Y.; Walter, K.; Spoerke, J.M.; O’Brien, C.; Huw, L.Y.; Hampton, G.M.; Lackner, M.R. Activating Mutations in PIK3CB Confer Resistance to PI3K Inhibition and Define a Novel Oncogenic Role for p110beta. Cancer Res. 2016, 76, 1193–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaudin, P.; Delanoue, R.; Davidson, I.; Silber, J.; Zider, A. TONDU (TDU), a novel human protein related to the product of vestigial (vg) gene of Drosophila melanogaster interacts with vertebrate TEF factors and substitutes for Vg function in wing formation. Development 1999, 126, 4807–4816. [Google Scholar]
- Zhou, Y.; Huang, T.; Cheng, A.S.; Yu, J.; Kang, W.; To, K.F. The TEAD Family and Its Oncogenic Role in Promoting Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 138. [Google Scholar] [CrossRef] [Green Version]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [Green Version]
- Lim, B.; Park, J.L.; Kim, H.J.; Park, Y.K.; Kim, J.H.; Sohn, H.A.; Noh, S.M.; Song, K.S.; Kim, W.H.; Kim, Y.S.; et al. Integrative genomics analysis reveals the multilevel dysregulation and oncogenic characteristics of TEAD4 in gastric cancer. Carcinogenesis 2014, 35, 1020–1027. [Google Scholar] [CrossRef]
- Zhang, W.; Gao, Y.; Li, P.; Shi, Z.; Guo, T.; Li, F.; Han, X.; Feng, Y.; Zheng, C.; Wang, Z.; et al. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2014, 24, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shen, H.; Withers, H.G.; Yang, N.; Denson, K.E.; Mussell, A.L.; Truskinovsky, A.; Fan, Q.; Gelman, I.H.; Frangou, C.; et al. VGLL4 Selectively Represses YAP-Dependent Gene Induction and Tumorigenic Phenotypes in Breast Cancer. Sci. Rep. 2017, 7, 6190. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Chan, S.W.; Lee, I.; Song, H.; Hong, W. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure 2012, 20, 1135–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pobbati, A.V.; Han, X.; Hung, A.W.; Weiguang, S.; Huda, N.; Chen, G.Y.; Kang, C.; Chia, C.S.; Luo, X.; Hong, W.; et al. Targeting the Central Pocket in Human Transcription Factor TEAD as a Potential Cancer Therapeutic Strategy. Structure 2015, 23, 2076–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilla, M.A.; Lopez-Garcia, M.A.; Atienza, M.R.; Rosa-Rosa, J.M.; Diaz-Martin, J.; Pecero, M.L.; Vieites, B.; Romero-Perez, L.; Benitez, J.; Calcabrini, A.; et al. VGLL1 expression is associated with a triple-negative basal-like phenotype in breast cancer. Endocr. Relat. Cancer 2014, 21, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.Z.; Yao, H.Q.; Zhu, S.Z.; Li, Q.Y.; Guo, G.H.; Yu, J. Expression levels of matrix metalloproteinase-9 in human gastric carcinoma. Oncol. Lett. 2015, 9, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Li, L.; Lin, J.Y.; Lin, H. Imbalance between expression of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in invasiveness and metastasis of human gastric carcinoma. World J. Gastroenterol. 2003, 9, 899–904. [Google Scholar] [CrossRef]
- Tran, P.; Nguyen, C.; Klempner, S.J. Targeting the Phosphatidylinositol-3-kinase Pathway in Gastric Cancer: Can Omics Improve Outcomes? Int. Neurourol. J. 2016, 20 (Suppl. S2), S131–S140. [Google Scholar] [CrossRef] [Green Version]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Mesrouze, Y.; Hau, J.C.; Erdmann, D.; Zimmermann, C.; Fontana, P.; Schmelzle, T.; Chene, P. The surprising features of the TEAD4-Vgll1 protein-protein interaction. Chembiochem 2014, 15, 537–542. [Google Scholar] [CrossRef]
- Pan, W.; Wang, Q.; Zhang, Y.; Zhang, N.; Qin, J.; Li, W.; Wang, J.; Wu, F.; Cao, L.; Xu, G. Verteporfin can Reverse the Paclitaxel Resistance Induced by YAP Over-Expression in HCT-8/T Cells without Photoactivation through Inhibiting YAP Expression. Cell Physiol. Biochem. 2016, 39, 481–490. [Google Scholar] [CrossRef]
- Hsu, P.C.; You, B.; Yang, Y.L.; Zhang, W.Q.; Wang, Y.C.; Xu, Z.; Dai, Y.; Liu, S.; Yang, C.T.; Li, H.; et al. YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget 2016, 7, 51922–51933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.K.; Nam, S.W.; Min, B.S.; Ban, H.S.; Paik, S.; Lee, K.; Im, J.Y.; Lee, Y.; Park, J.T.; Kim, S.Y.; et al. Bcl-2-dependent synthetic lethal interaction of the IDF-11774 with the V0 subunit C of vacuolar ATPase (ATP6V0C) in colorectal cancer. Br. J. Cancer 2018, 119, 1347–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelm, J.M.; Timmins, N.E.; Brown, C.J.; Fussenegger, M.; Nielsen, L.K. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol. Bioeng. 2003, 83, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Stine, Z.E.; Nguyen, C.; Afzal, J.; Sun, P.; Hamaker, M.; Siegel, N.M.; Gouw, A.M.; Kang, B.H.; Yu, S.H.; et al. Tumorigenicity of hypoxic respiring cancer cells revealed by a hypoxia-cell cycle dual reporter. Proc. Natl. Acad. Sci. USA 2014, 111, 12486–12491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, Y.; Li, Q.; Kazarov, A.R.; Epardaud, M.; Elpek, K.; Turley, S.J.; Hemler, M.E. Diminished metastasis in tetraspanin CD151-knockout mice. Blood 2011, 118, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.K.; Im, J.Y.; Han, G.; Lee, W.J.; Won, K.J.; Chung, K.S.; Lee, K.; Ban, H.S.; Song, K.; Won, M. p300 cooperates with c-Jun and PARP-1 at the p300 binding site to activate RhoB transcription in NSC126188-mediated apoptosis. Biochim. Biophys. Acta 2014, 1839, 364–373. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.-K.; Cheong, J.-H.; Im, J.-Y.; Ban, H.S.; Kim, S.-K.; Kang, M.-J.; Lee, J.; Kim, S.-Y.; Park, K.-C.; Paik, S.; et al. PI3K/AKT/β-Catenin Signaling Regulates Vestigial-Like 1 Which Predicts Poor Prognosis and Enhances Malignant Phenotype in Gastric Cancer. Cancers 2019, 11, 1923. https://doi.org/10.3390/cancers11121923
Kim B-K, Cheong J-H, Im J-Y, Ban HS, Kim S-K, Kang M-J, Lee J, Kim S-Y, Park K-C, Paik S, et al. PI3K/AKT/β-Catenin Signaling Regulates Vestigial-Like 1 Which Predicts Poor Prognosis and Enhances Malignant Phenotype in Gastric Cancer. Cancers. 2019; 11(12):1923. https://doi.org/10.3390/cancers11121923
Chicago/Turabian StyleKim, Bo-Kyung, Jae-Ho Cheong, Joo-Young Im, Hyun Seung Ban, Seon-Kyu Kim, Mi-Jung Kang, Jungwoon Lee, Seon-Young Kim, Kyung-Chan Park, Soonmyung Paik, and et al. 2019. "PI3K/AKT/β-Catenin Signaling Regulates Vestigial-Like 1 Which Predicts Poor Prognosis and Enhances Malignant Phenotype in Gastric Cancer" Cancers 11, no. 12: 1923. https://doi.org/10.3390/cancers11121923
APA StyleKim, B.-K., Cheong, J.-H., Im, J.-Y., Ban, H. S., Kim, S.-K., Kang, M.-J., Lee, J., Kim, S.-Y., Park, K.-C., Paik, S., & Won, M. (2019). PI3K/AKT/β-Catenin Signaling Regulates Vestigial-Like 1 Which Predicts Poor Prognosis and Enhances Malignant Phenotype in Gastric Cancer. Cancers, 11(12), 1923. https://doi.org/10.3390/cancers11121923