Anti-Tumor Effect of Inhibition of DNA Damage Response Proteins, ATM and ATR, in Endometrial Cancer Cells


 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
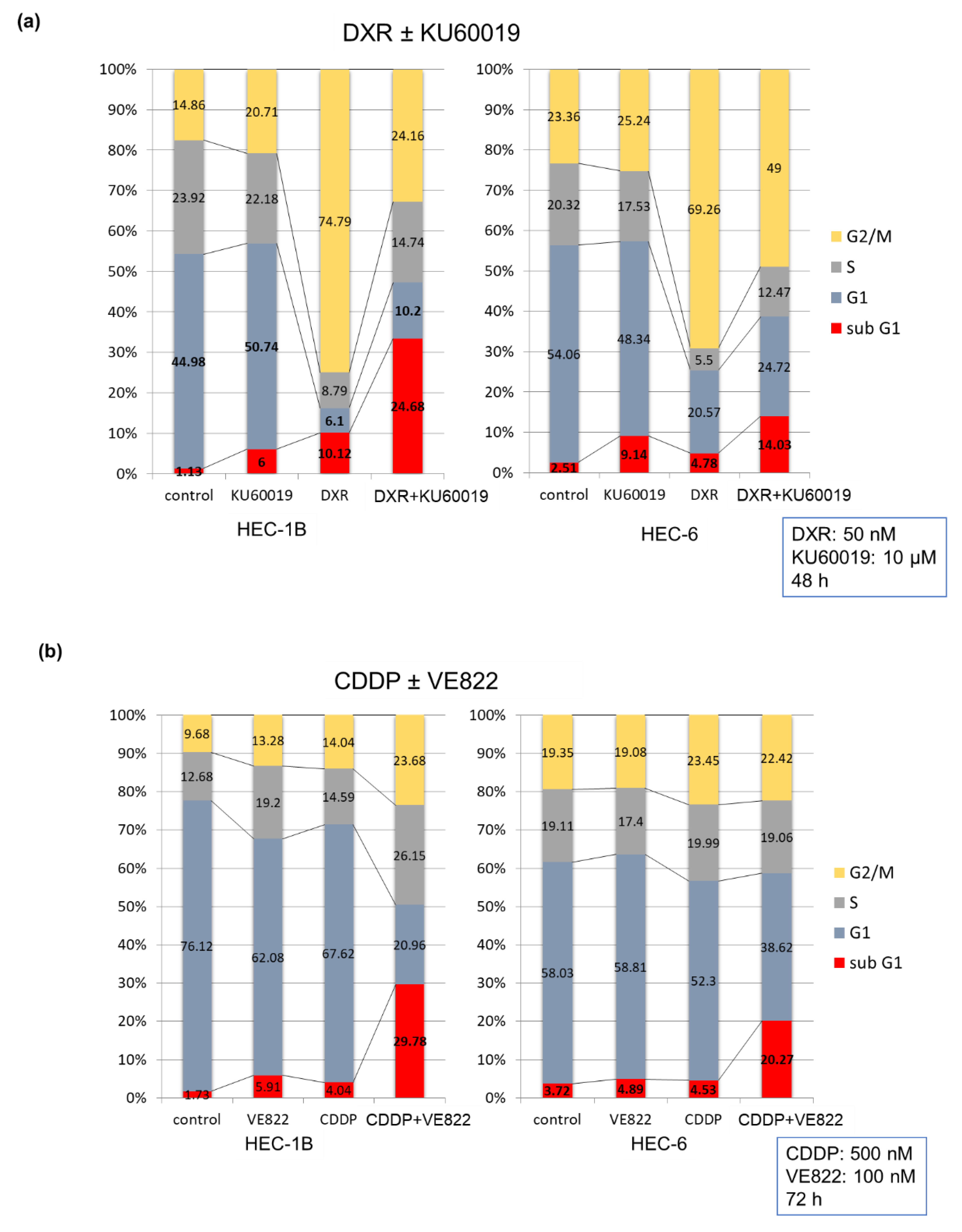
2.1. The Effect of ATR Inhibitor or ATM Inhibitor Combined with DXR or CDDP in Endometrial Cancer Cells
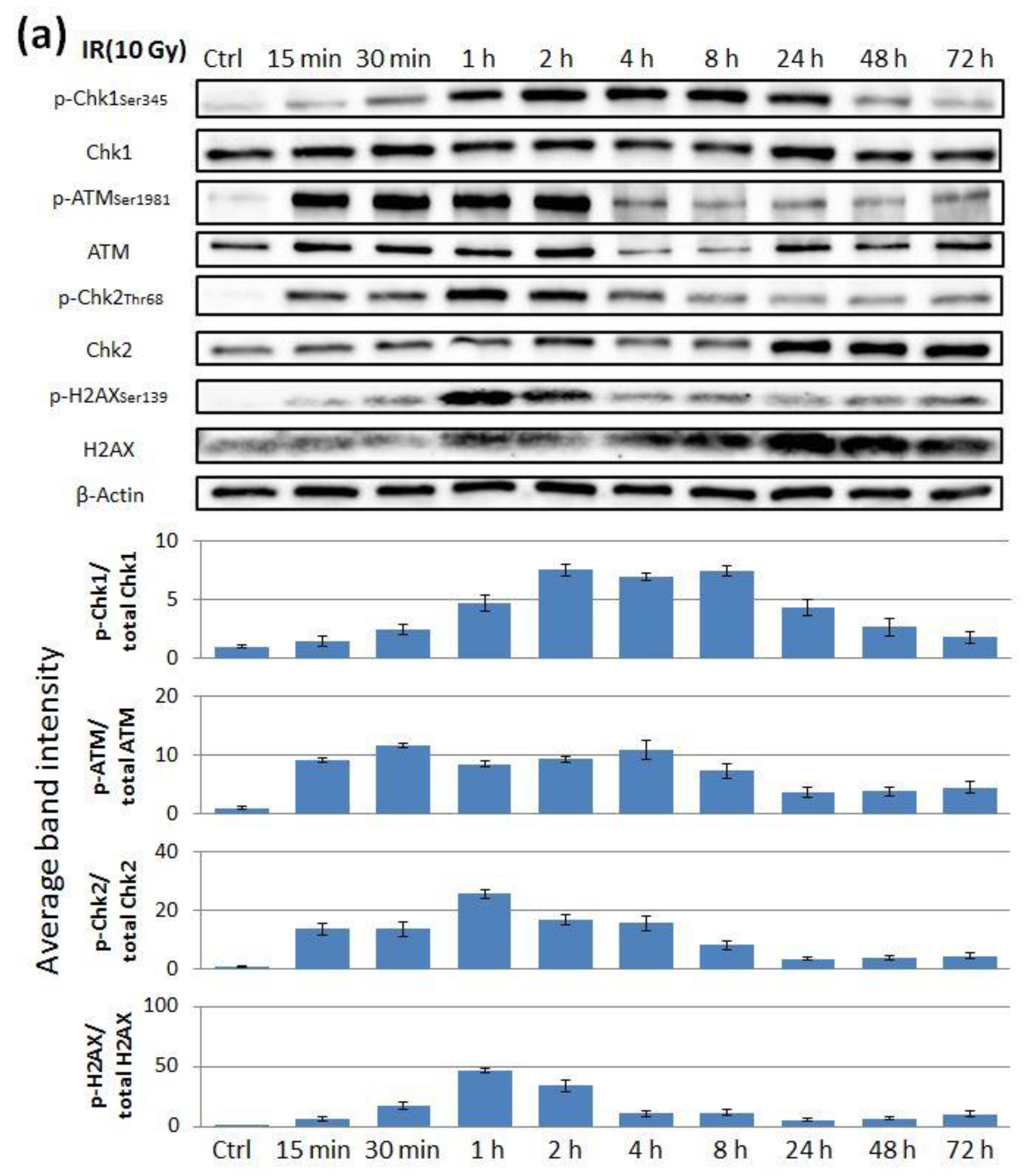
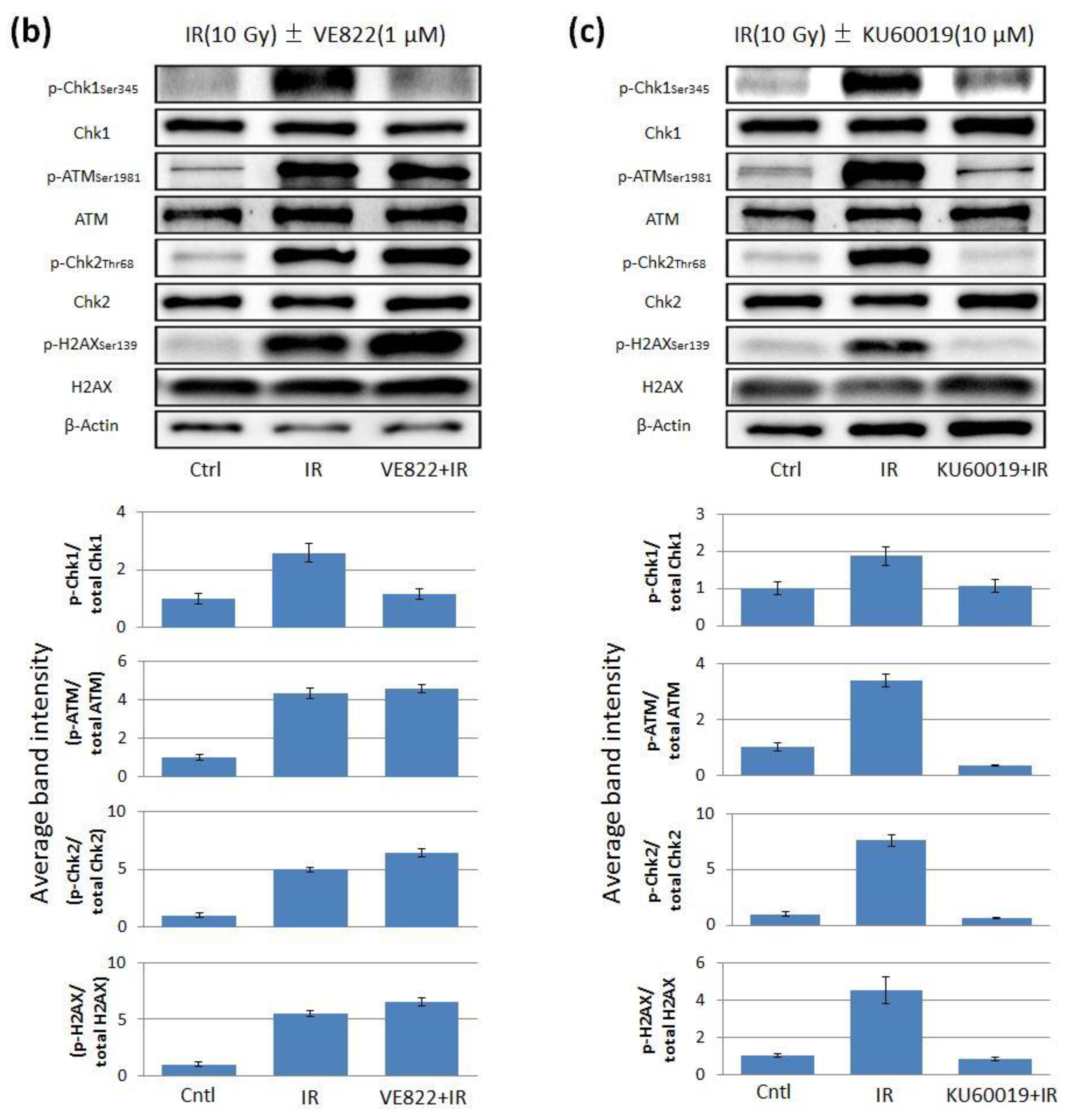
2.2. The Effect of ATR Inhibitor or ATM Inhibitor Combined with Irradiation in Endometrial Cancer Cells
2.3. The Effect of the Combination of ATR Inhibitor and Chk1 Inhibitor in Endometrial Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Viability Assay
4.3. Clonogenic Assay
4.4. Immunoblotting
4.5. Cell Cycle Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Buring, J.E.; Bain, C.J.; Ehrmann, R.L. Conjugated estrogen use and risk of endometrial cancer. Am. J. Epidemiol. 1986, 124, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Brinton, L.A.; Lacey, J.V., Jr.; Sherman, M.E. Epidemiology of Gynecologic Cancers. Principles and Practice of Gynecologic Oncology, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005; pp. 3–32. [Google Scholar]
- Ko, E.M.; Walter, P.; Clark, L.; Jackson, A.; Franasiak, J.; Bolac, C.; Havrilesky, L.; Secord, A.A.; Moore, D.T.; Gehrig, P.A.; et al. The complex triad of obesity, diabetes and race in Type I and II endometrial cancers: Prevalence and prognostic significance. Gynecol. Oncol. 2014, 133, 28–32. [Google Scholar] [CrossRef]
- The Research Group for Population-Based Cancer Registration in Japan. Cancer incidence and incidence rates in Japan in 1998: Estimates based on data from 12 population-based cancer registries. JPN J. Clin. Oncol. 2003, 33, 241–245. [Google Scholar] [CrossRef][Green Version]
- Randall, M.E.; Filiaci, V.L.; Muss, H.; Spirtos, N.M.; Mannel, R.S.; Fowler, J.; Thigpen, J.T.; Benda, J.A. Randomized phase III trial of whole-abdominal irradiation versus doxorubicin and cisplatin chemotherapy in advanced endometrial carcinoma: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2006, 24, 36–44. [Google Scholar] [CrossRef]
- Colombo, N.; Preti, E.; Landoni, F.; Carinelli, S.; Colombo, A.; Marini, C.; Sessa, C.; ESMO Guidelines Working Group. Endometrial cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2011, 22 (Suppl. 6), vi35–vi39. [Google Scholar] [CrossRef]
- Wright, J.D.; Medel, N.I.B.; Sehouli, J.; Fujiwara, K.; Herzog, T.J. Contemporary management of endometrial cancer. Lancet 2012, 379, 1352–1360. [Google Scholar] [CrossRef]
- Morrow, C.P.; Bundy, B.N.; Kurman, R.J.; Creasman, W.T.; Heller, P.; Homesley, H.D.; Graham, J.E. Relationship between surgical-pathological risk factors and outcome in clinical stage I and II carcinoma of the endometrium: A Gynecologic Oncology Group study. Gynecol. Oncol. 1991, 40, 55–65. [Google Scholar] [CrossRef]
- Creutzberg, C.L.; van Putten, W.L.; Koper, P.C.; Lybeert, M.L.; Jobsen, J.J.; Wárlám-Rodenhuis, C.C.; De Winter, K.A.; Lutgens, L.C.; van den Bergh, A.C.; van de Steen-Banasik, E.; et al. Surgery and postoperative radiotherapy versus surgery alone for patients with stage-1 endometrial carcinoma: Multicentre randomised trial. PORTEC Study Group. Post Operative Radiation Therapy in Endometrial Carcinoma. Lancet 2000, 355, 1404–1411. [Google Scholar] [CrossRef]
- Hosoya, N.; Miyagawa, K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Okada, J.; Timmerman, L.; Rodriguez-Viciana, P.; Stokoe, D.; Shoji, K.; Taketani, Y.; Kuramoto, H.; Knight, Z.A.; Shokat, K.M.; et al. PIK3CA cooperates with other phosphatidylinositol 3’-kinase pathway mutations to effect oncogenic transformation. Cancer Res. 2008, 68, 8127–8136. [Google Scholar] [CrossRef]
- Griffith, E.; Walker, S.; Martin, C.A.; Vagnarelli, P.; Stiff, T.; Vernay, B.; Al Sanna, N.; Saggar, A.; Hamel, B.; Earnshaw, W.C.; et al. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat. Genet. 2008, 40, 232–236. [Google Scholar] [CrossRef]
- Zhou, B.B.; Bartek, J. Targeting the checkpoint kinases: Chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–225. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Lavin, M.F.; Shiloh, Y. The genetic defect in ataxia-telangiectasia. Annu. Rev. Immunol. 1997, 15, 177–202. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Teng, P.N.; Bateman, N.W.; Darcy, K.M.; Hamilton, C.A.; Maxwell, G.L.; Bakkenist, C.J.; Conrads, T.P. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol. Oncol. 2015, 136, 554–561. [Google Scholar] [CrossRef]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Angele, S.; Treilleux, I.; Bremond, A.; Taniere, P.; Hall, J. Altered expression of DNA double-strand break detection and repair proteins in breast carcinomas. Histopathology 2003, 43, 347–353. [Google Scholar] [CrossRef]
- Ai, L.; Vo, Q.N.; Zuo, C.; Li, L.; Ling, W.; Suen, J.Y.; Hanna, E.; Brown, K.D.; Fan, C.Y. Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: Promoter hypermethylation with clinical correlation in 100 cases. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 150–156. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, M.J.; Lee, J.Y.; Lee, S.M.; Choi, J.E.; Lee, S.Y.; Park, J.Y. Epigenetic inactivation of checkpoint kinase 2 gene in non-small cell lung cancer and its relationship with clinicopathological features. Lung Cancer 2009, 65, 247–250. [Google Scholar] [CrossRef]
- Sullivan, A.; Yuille, M.; Repellin, C.; Reddy, A.; Reelfs, O.; Bell, A.; Dunne, B.; Gusterson, B.A.; Osin, P.; Farrell, P.J.; et al. Concomitant inactivation of p53 and Chk2 in breast cancer. Oncogene 2002, 21, 1316–1324. [Google Scholar] [CrossRef][Green Version]
- Sanjiv, K.; Hagenkort, A.; Calderon-Montano, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R.V.; Schultz, N.; Scobie, M.; et al. Cancer-specific synthetic lethality between ATR and CHK1 kinase activities. Cell Rep. 2016, 14, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Lee, C.H.; Chung, J.H. The hCds1 (Chk2)-FHA domain is essential for a chain of phosphorylation events on hCds1 that is induced by ionizing radiation. J. Biol. Chem. 2001, 276, 30537–30541. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Hammond, E.M.; Brunner, T.B.; McKenna, W.G.; Muschel, R.J. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat. Rev. 2014, 40, 109–117. [Google Scholar] [CrossRef]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Zighelboim, I.; Schmidt, A.P.; Gao, F.; Thaker, P.H.; Powell, M.A.; Rader, J.S.; Gibb, R.K.; Mutch, D.G.; Goodfellow, P.J. ATR mutation in endometrioid endometrial cancer is associated with poor clinical outcomes. J. Clin. Oncol. 2009, 27, 3091–3096. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Wang, C.; Zhang, Z.; Luo, X.; Ning, C.; Yu, Y.; Feng, Y.; Gu, C.; Chen, X. ATM may be a protective factor in endometrial carcinogenesis with the progesterone pathway. Tumour. Biol. 2015, 36, 1529–1537. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeuchi, M.; Tanikawa, M.; Nagasaka, K.; Oda, K.; Kawata, Y.; Oki, S.; Agapiti, C.; Sone, K.; Miyagawa, Y.; Hiraike, H.; et al. Anti-Tumor Effect of Inhibition of DNA Damage Response Proteins, ATM and ATR, in Endometrial Cancer Cells. Cancers 2019, 11, 1913. https://doi.org/10.3390/cancers11121913
Takeuchi M, Tanikawa M, Nagasaka K, Oda K, Kawata Y, Oki S, Agapiti C, Sone K, Miyagawa Y, Hiraike H, et al. Anti-Tumor Effect of Inhibition of DNA Damage Response Proteins, ATM and ATR, in Endometrial Cancer Cells. Cancers. 2019; 11(12):1913. https://doi.org/10.3390/cancers11121913
Chicago/Turabian StyleTakeuchi, Makoto, Michihiro Tanikawa, Kazunori Nagasaka, Katsutoshi Oda, Yoshiko Kawata, Shinya Oki, Chuwa Agapiti, Kenbun Sone, Yuko Miyagawa, Haruko Hiraike, and et al. 2019. "Anti-Tumor Effect of Inhibition of DNA Damage Response Proteins, ATM and ATR, in Endometrial Cancer Cells" Cancers 11, no. 12: 1913. https://doi.org/10.3390/cancers11121913
APA StyleTakeuchi, M., Tanikawa, M., Nagasaka, K., Oda, K., Kawata, Y., Oki, S., Agapiti, C., Sone, K., Miyagawa, Y., Hiraike, H., Wada-Hiraike, O., Kuramoto, H., Ayabe, T., Osuga, Y., & Fujii, T. (2019). Anti-Tumor Effect of Inhibition of DNA Damage Response Proteins, ATM and ATR, in Endometrial Cancer Cells. Cancers, 11(12), 1913. https://doi.org/10.3390/cancers11121913