Proteogenomics of Colorectal Cancer Liver Metastases: Complementing Precision Oncology with Phenotypic Data

 ,
, 



Abstract
1. Introduction
2. Results
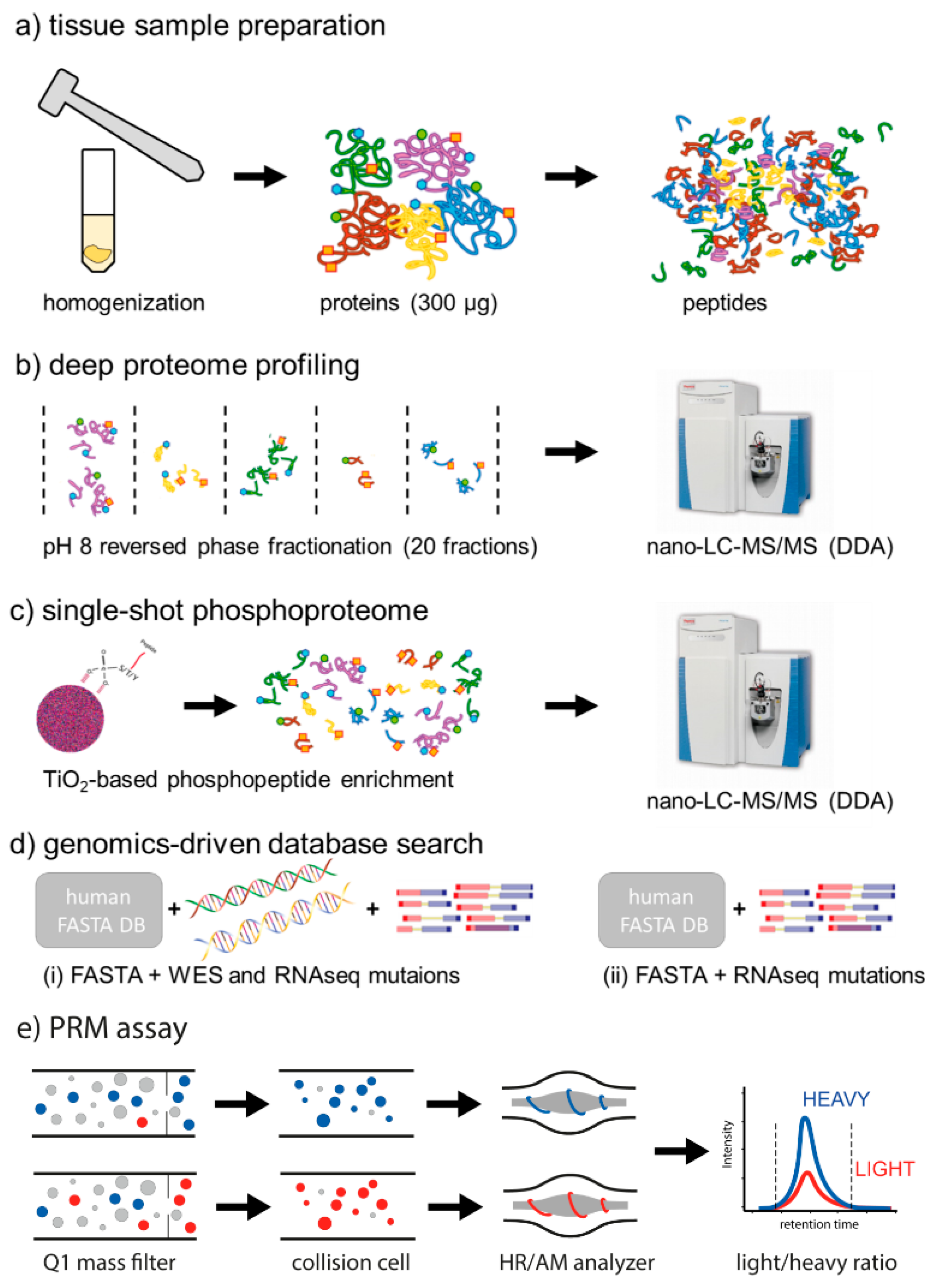
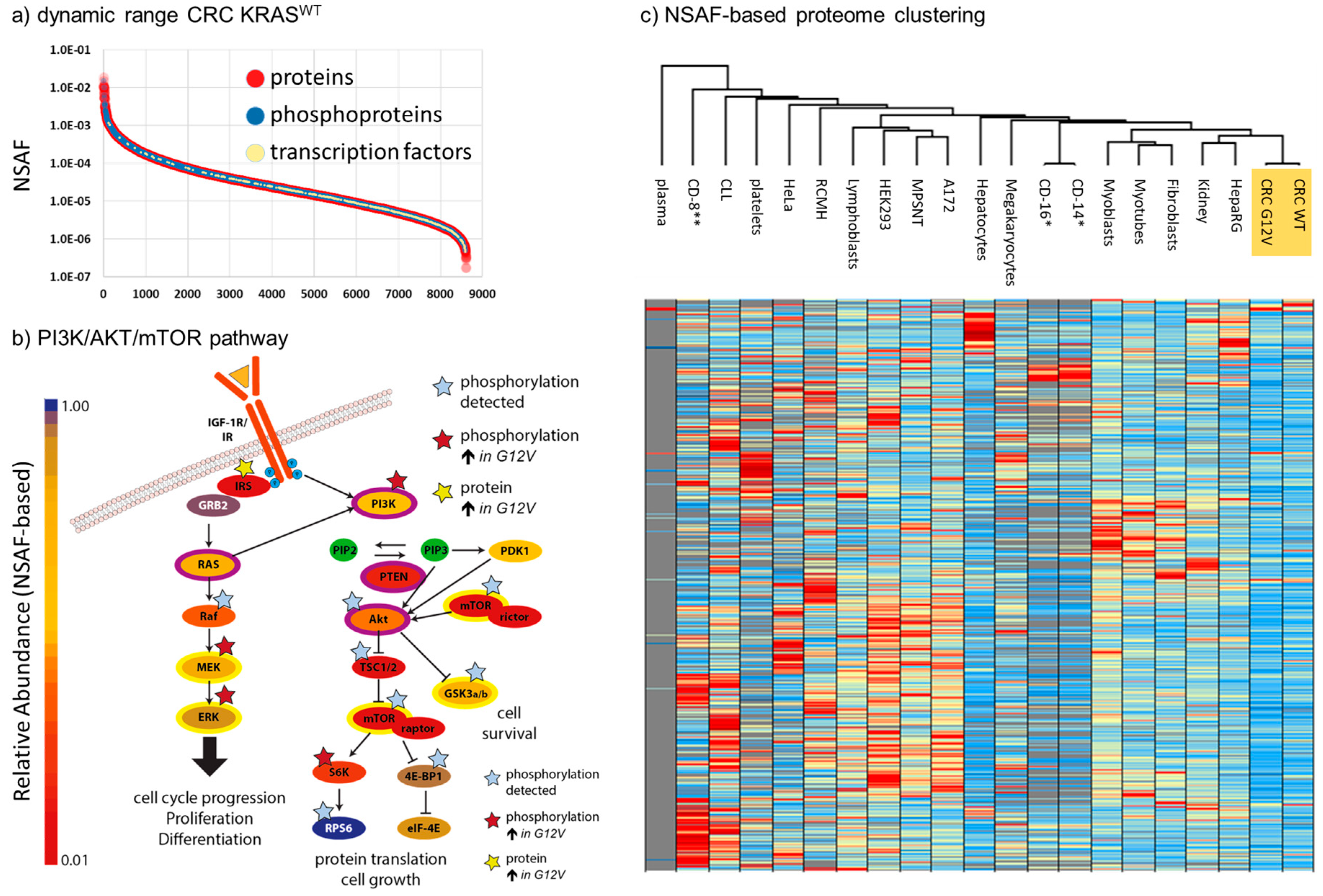
2.1. Depth of the Proteome
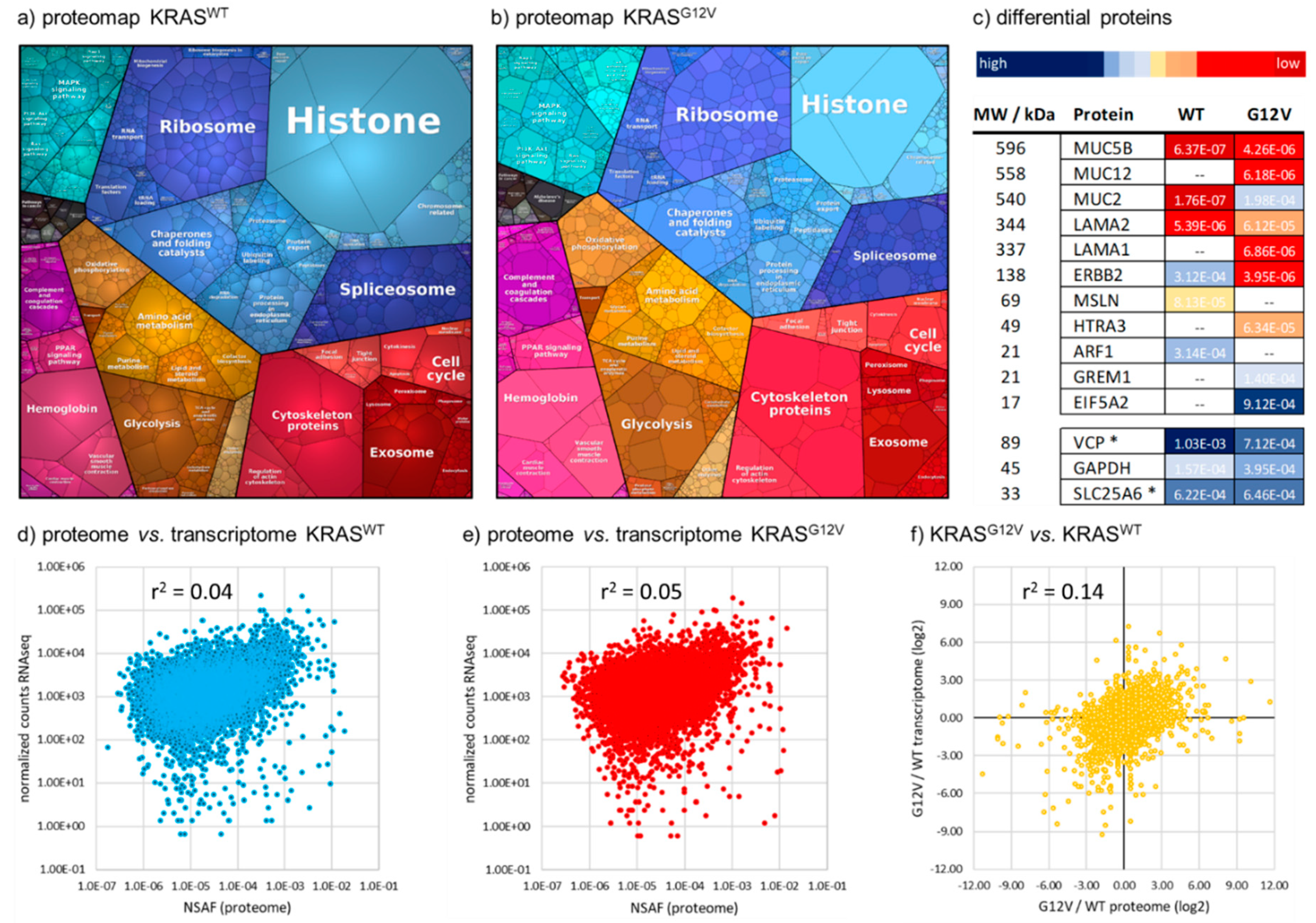
2.2. Quantitative Comparison of KRASG12V and KRASWT Tumors
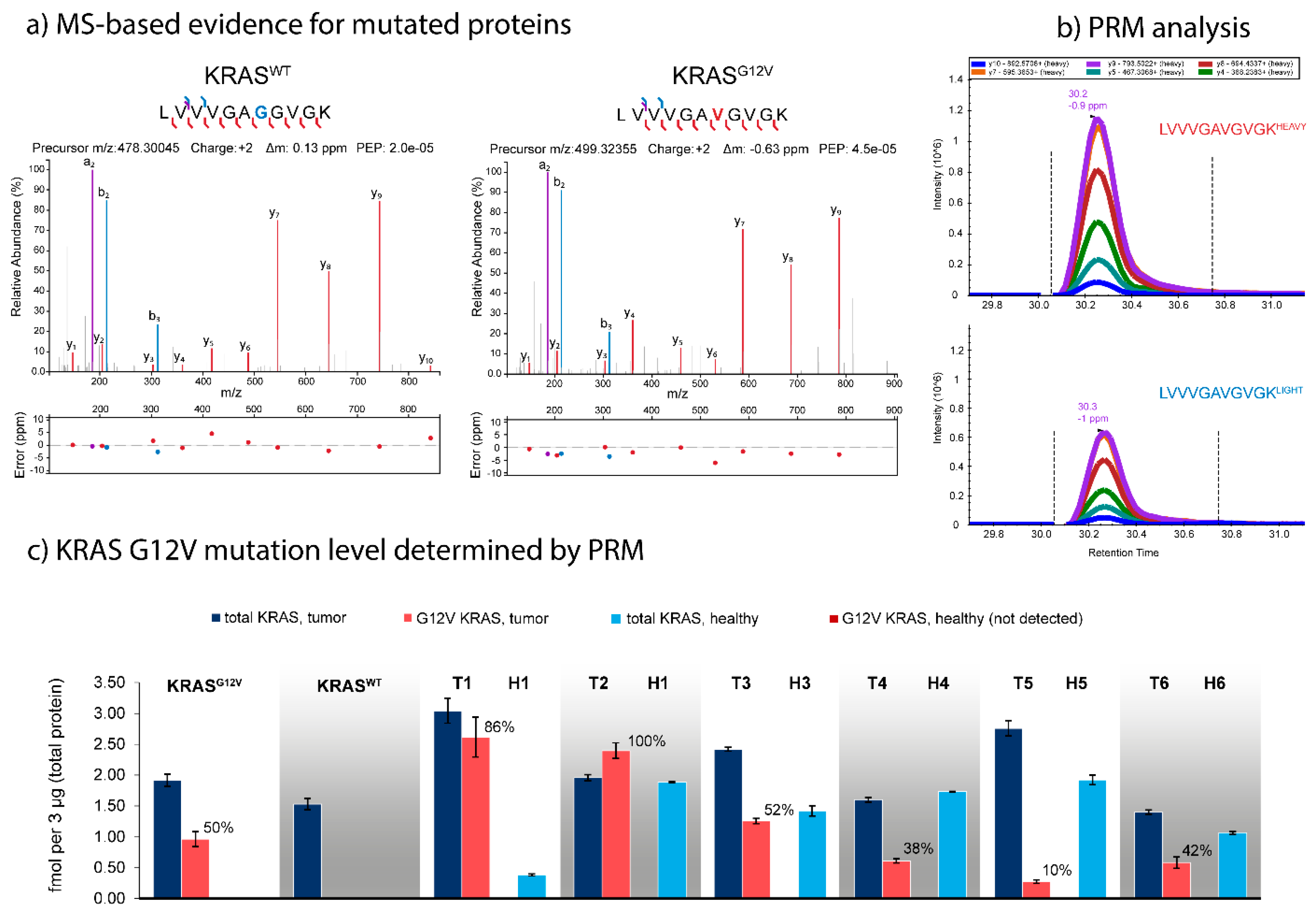
2.3. Identification and Quantification of SNV Predicted from the WES and RNAseq Data
2.4. Robustness of NSAF-Based Relative Quantification
3. Discussion
4. Materials and Methods
4.1. Experimental Design and Statistical Rationale
4.2. Materials
4.3. Sampling
4.4. WES and RNAseq Data Generation
4.5. Sample Preparation and Discovery Proteomics
4.5.1. Tissue Homogenization and Protein Extraction
4.5.2. Protein Concentration Estimation and Proteolytic Digestion
4.5.3. Peptide Fractionation for Global Proteome Analysis
4.5.4. Phosphopeptide Enrichment for Phosphoproteome Analysis
4.5.5. Nano-LC-MS/MS in Data Dependent Acquisition Mode
4.6. Discovery Data Search and Analysis
4.6.1. Database generation
4.6.2. Database Search Strategies
4.6.3. Quantitative Comparison of the Deep Proteome Data
4.7. Development of Parallel Reaction Monitoring Assays for Selected Mutations
4.7.1. Mutation Selection
4.7.2. Synthesis, Purification, and Quantification of Stable Isotope Labeled Standard (SIS) Peptides
4.7.3. Nano-LC-MS/MS in PRM Mode
4.7.4. PRM Development and Analysis
5. Conclusions
Data and Materials Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BCA | bicinchoninic acid assay |
| CRC | colorectal cancer |
| OCT | Optimal cutting temperature compound |
| PRM | parallel reaction monitoring |
| RNAseq | RNA sequencing |
| SPE | solid phase extraction |
| WES | whole exome sequencing |
References
- American Institute for Cancer Research. Colorectal Cancer Statistics. Colorectal Cancer is the Third Most Common Cancer Worldwide. Available online: https://www.wcrf.org/dietandcancer/cancer-trends/colorectal-cancer-statistics (accessed on 29 November 2019).
- World Helalth Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 29 November 2019).
- Martínez, M.E. Primary prevention of colorectal cancer: Lifestyle, nutrition, exercise. Recent Results Cancer Res. 2005, 166, 177–211. [Google Scholar] [PubMed]
- Van Cutsem, E.; Nordlinger, B.; Adam, R.; Köhne, C.H.; Pozzo, C.; Poston, G.; Ychou, M.; Rougier, P.; European Colorectal Metastases Treatment Group. Towards a pan-European consensus on the treatment of patients with colorectal liver metastases. Eur. J. Cancer 2006, 42, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- American Chemical Society. Survival Rates for Colorectal Cancer. Available online: http://www.cancer.org/cancer/colon-rectal-cancer/detection-diagnosis-staging/survival-rates.html (accessed on 29 November 2019).
- Cohen, R.B. Epidermal growth factor receptor as a therapeutic target in colorectal cancer. Clin. Colorectal Cancer 2003, 2, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Pabla, B.; Bissonnette, M.; Konda, V.J. Colon cancer and the epidermal growth factor receptor: Current treatment paradigms, the importance of diet, and the role of chemoprevention. World J. Clin. Oncol. 2015, 6, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Mitchell, E.; Chidiac, T.; Scroggin, C.; Hagenstad, C.; Spigel, D.; Marshall, J.; Cohn, A.; McCollum, D.; Stella, P.; et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 672–680. [Google Scholar] [CrossRef]
- Gallagher, D.J.; Kemeny, N. Metastatic colorectal cancer: From improved survival to potential cure. Oncology 2010, 78, 237–248. [Google Scholar] [CrossRef]
- Kato, S.; Okamura, R.; Mareboina, M.; Lee, S.; Goodman, A.; Patel, S.P.; Fanta, P.T.; Schwab, R.B.; Vu, P.; Raymond, V.M.; et al. Revisiting Epidermal Growth Factor Receptor (EGFR) Amplification as a Target for Anti-EGFR Therapy: Analysis of Cell-Free Circulating Tumor DNA in Patients With Advanced Malignancies. JCO Precis. Oncol. 2019, 3, 1–14. [Google Scholar] [CrossRef]
- Lièvre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Côté, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- Mittmann, N.; Au, H.J.; Tu, D.; O’Callaghan, C.J.; Isogai, P.K.; Karapetis, C.S.; Zalcberg, J.R.; Evans, W.K.; Moore, M.J.; Siddiqui, J.; et al. Prospective cost-effectiveness analysis of cetuximab in metastatic colorectal cancer: Evaluation of National Cancer Institute of Canada Clinical Trials Group CO.17 trial. J. Natl. Cancer Inst. 2009, 101, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Cushman, S.M.; Jiang, C.; Hatch, A.J.; Shterev, I.; Sibley, A.B.; Niedzwiecki, D.; Venook, A.P.; Owzar, K.; Hurwitz, H.I.; Nixon, A.B. Gene expression markers of efficacy and resistance to cetuximab treatment in metastatic colorectal cancer: Results from CALGB 80203 (Alliance). Clin. Cancer Res. 2015, 21, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Carbonell, W.S.; Hu, Y.L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef]
- Mnatsakanyan, R.; Shema, G.; Basik, M.; Batist, G.; Borchers, C.H.; Sickmann, A.; Zahedi, R.P. Detecting post-translational modification signatures as potential biomarkers in clinical mass spectrometry. Expert Rev. Proteom. 2018, 15, 515–535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.; Hickman, J.A. Limits to Precision Cancer Medicine. N. Engl. J. Med. 2017, 376, 96–97. [Google Scholar] [CrossRef]
- Zhu, Y.; Orre, L.M.; Johansson, H.J.; Huss, M.; Boekel, J.; Vesterlund, M.; Fernandez-Woodbridge, A.; Branca, R.M.M.; Lehtiö, J. Discovery of coding regions in the human genome by integrated proteogenomics analysis workflow. Nat. Commun. 2018, 9, e903. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- Huang, K.L.; Li, S.; Mertins, P.; Cao, S.; Gunawardena, H.P.; Ruggles, K.V.; Mani, D.R.; Clauser, K.R.; Tanioka, M.; Usary, J.; et al. Proteogenomic integration reveals therapeutic targets in breast cancer xenografts. Nat. Commun. 2017, 8, e14864. [Google Scholar] [CrossRef]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049. [Google Scholar] [CrossRef]
- Paoletti, A.C.; Parmely, T.J.; Tomomori-Sato, C.; Sato, S.; Zhu, D.; Conaway, R.C.; Conaway, J.W.; Florens, L.; Washburn, M.P. Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. USA 2006, 103, 18928–18933. [Google Scholar] [CrossRef] [PubMed]
- Bubis, J.A.; Levitsky, L.I.; Ivanov, M.V.; Tarasova, I.A.; Gorshkov, M.V. Comparative evaluation of label-free quantification methods for shotgun proteomics. Rapid Commun. Mass Spectrom. 2017, 31, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Eid, S.; Turk, S.; Volkamer, A.; Rippmann, F.; Fulle, S. KinMap: A web-based tool for interactive navigation through human kinome data. BMC Bioinform. 2017, 18, e16. [Google Scholar] [CrossRef]
- Liberti, S.; Sacco, F.; Calderone, A.; Perfetto, L.; Iannuccelli, M.; Panni, S.; Santonico, E.; Palma, A.; Nardozza, A.P.; Castagnoli, L.; et al. HuPho: The human phosphatase portal. FEBS J. 2013, 280, 379–387. [Google Scholar] [CrossRef]
- Pérez-Silva, J.G.; Español, Y.; Velasco, G.; Quesada, V. The Degradome database: Expanding roles of mammalian proteases in life and disease. Nucleic Acids Res. 2016, 44, 351–355. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, 649–655. [Google Scholar] [CrossRef]
- Kollipara, L.; Buchkremer, S.; Weis, J.; Brauers, E.; Hoss, M.; Rütten, S.; Caviedes, P.; Zahedi, R.P.; Roos, A. Proteome Profiling and Ultrastructural Characterization of the Human RCMH Cell Line: Myoblastic Properties and Suitability for Myopathological Studies. J. Proteome Res. 2016, 15, 945–955. [Google Scholar] [CrossRef]
- Segura, V.; Valero, M.L.; Cantero, L.; Muñoz, J.; Zarzuela, E.; García, F.; Aloria, K.; Beaskoetxea, J.; Arizmendi, J.M.; Navajas, R.; et al. In-Depth Proteomic Characterization of Classical and Non-Classical Monocyte Subsets. Proteomes 2018, 6, 8. [Google Scholar] [CrossRef]
- van Aalderen, M.C.; van den Biggelaar, M.; Remmerswaal, E.B.M.; van Alphen, F.P.J.; Meijer, A.B.; Ten Berge, I.J.M.; van Lier, R.A.W. Label-free Analysis of CD8+ T Cell Subset Proteomes Supports a Progressive Differentiation Model of Human-Virus-Specific T Cells. Cell Rep. 2017, 19, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual account of protein investment in cellular functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jin, S.; Lu, H.; Mi, S.; Shao, W.; Zuo, X.; Yin, H.; Zeng, S.; Shimamoto, F.; Qi, G. Expression of survivin, MUC2 and MUC5 in colorectal cancer and their association with clinicopathological characteristics. Oncol. Lett. 2017, 14, 1011–1016. [Google Scholar] [CrossRef]
- Matsuyama, T.; Ishikawa, T.; Mogushi, K.; Yoshida, T.; Iida, S.; Uetake, H.; Mizushima, H.; Tanaka, H.; Sugihara, K. MUC12 mRNA expression is an independent marker of prognosis in stage II and stage III colorectal cancer. Int. J. Cancer 2010, 127, 2292–2299. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Jiang, Z.; Stawiski, E.W.; Gnad, F.; Liu, J.; Mayba, O.; Du, P.; Diao, J.; Johnson, S.; Wong, K.F.; et al. Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol. 2014, 15, e436. [Google Scholar] [CrossRef]
- Rao, C.V.; Asch, A.S.; Yamada, H.Y. Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. Carcinogenesis 2017, 38, 2–11. [Google Scholar] [CrossRef]
- Casalou, C.; Faustino, A.; Barral, D.C. Arf proteins in cancer cell migration. Small Gtpases 2016, 7, 270–282. [Google Scholar] [CrossRef]
- Davis, J.E.; Xie, X.; Guo, J.; Huang, W.; Chu, W.M.; Huang, S.; Teng, Y.; Wu, G. ARF1 promotes prostate tumorigenesis via targeting oncogenic MAPK signaling. Oncotarget 2016, 7, 39834–39845. [Google Scholar] [CrossRef]
- Ross, J.S.; Fakih, M.; Ali, S.M.; Elvin, J.A.; Schrock, A.B.; Suh, J.; Vergilio, J.A.; Ramkissoon, S.; Severson, E.; Daniel, S.; et al. Targeting HER2 in colorectal cancer: The landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer 2018, 124, 1358–1373. [Google Scholar] [CrossRef]
- Jang, B.G.; Kim, H.S.; Chang, W.Y.; Bae, J.M.; Oh, H.J.; Wen, X.; Jeong, S.; Cho, N.Y.; Kim, W.H.; Kang, G.H. Prognostic significance of stromal GREM1 expression in colorectal cancer. Hum. Pathol. 2017, 62, 56–65. [Google Scholar] [CrossRef]
- Bao, Y.; Lu, Y.; Wang, X.; Feng, W.; Sun, X.; Guo, H.; Tang, C.; Zhang, X.; Shi, Q.; Yu, H. Eukaryotic translation initiation factor 5A2 (eIF5A2) regulates chemoresistance in colorectal cancer through epithelial mesenchymal transition. Cancer Cell Int. 2015, 15, e109. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I. Proteogenomics: Concepts, applications and computational strategies. Nat. Methods. 2014, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.L.; Wu, J.; Zhou, Y.; Fan, J.H. Increased Sushi repeat-containing protein X-linked 2 is associated with progression of colorectal cancer. Med. Oncol. 2015, 32, e99. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, e1260419. [Google Scholar] [CrossRef]
- Brademan, D.R.; Riley, N.M.; Kwiecien, N.W.; Coon, J.J. Interactive Peptide Spectral Annotator: A Versatile Web-Based Tool for Proteomic Applications. Mol. Cell. Proteom. 2019. [Google Scholar] [CrossRef]
- Marquart, J.; Chen, E.Y.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Benefit From Genome-Driven Oncology. JAMA Oncol. 2018, 4, 1093–1098. [Google Scholar] [CrossRef]
- Aguilar-Mahecha, A.; Lafleur, J.; Pelmus, M.; Seguin, C.; Lan, C.; Discepola, F.; Kovacina, B.; Christodoulopoulos, R.; Salvucci, O.; Mihalcioiu, C.; et al. The identification of challenges in tissue collection for biomarker studies: The Q-CROC-03 neoadjuvant breast cancer translational trial experience. Mod. Pathol. 2017, 30, 1567–1576. [Google Scholar] [CrossRef][Green Version]
- Diaz, Z.; Aguilar-Mahecha, A.; Paquet, E.R.; Basik, M.; Orain, M.; Camlioglu, E.; Constantin, A.; Benlimame, N.; Bachvarov, D.; Jannot, G.; et al. Next-generation biobanking of metastases to enable multidimensional molecular profiling in personalized medicine. Mod. Pathol. 2013, 26, 1413–1424. [Google Scholar] [CrossRef]
- Basik, M.; Aguilar-Mahecha, A.; Rousseau, C.; Diaz, Z.; Tejpar, S.; Spatz, A.; Greenwood, C.M.; Batist, G. Biopsies: Next-generation biospecimens for tailoring therapy. Nat. Rev. Clin. Oncol. 2013, 10, 437–450. [Google Scholar] [CrossRef]
- Pagel, O.; Loroch, S.; Sickmann, A.; Zahedi, R.P. Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev. Proteom. 2015, 12, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Rusch, M.; Nakitandwe, J.; Shurtleff, S.; Newman, S.; Zhang, Z.; Edmonson, M.N.; Parker, M.; Jiao, Y.; Ma, X.; Liu, Y.; et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat. Commun. 2018, 9, e3962. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, e11. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Schumbrutzki, C.; Wortelkamp, S.; Sickmann, A.; Zahedi, R.P. Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J. Prot. 2012, 75, 1454–1462. [Google Scholar] [CrossRef]
- Engholm-Keller, K.; Birck, P.; Størling, J.; Pociot, F.; Mandrup-Poulsen, T.; Larsen, M.R. TiSH—A robust and sensitive global phosphoproteomics strategy employing a combination of TiO2, SIMAC, and HILIC. J. Prot. 2012, 75, 5749–5761. [Google Scholar] [CrossRef] [PubMed]
- Gonczarowska-Jorge, H.; Loroch, S.; Dell’Aica, M.; Sickmann, A.; Roos, A.; Zahedi, R.P. Quantifying Missing (Phospho)Proteome Regions with the Broad-Specificity Protease Subtilisin. Anal. Chem. 2017, 89, 13137–13145. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Käll, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, e923. [Google Scholar] [CrossRef]
- Taus, T.; Köcher, T.; Pichler, P.; Paschke, C.; Schmidt, A.; Henrich, C.; Mechtler, K. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 2011, 10, 5354–5362. [Google Scholar] [CrossRef]
- Vaudel, M.; Barsnes, H.; Berven, F.S.; Sickmann, A.; Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 2011, 11, 996–999. [Google Scholar] [CrossRef]
- Bioconductor Open Source Software for Bioinformatics. Utilities for Exploration and Assessment of Confidence of LC-MSn Proteomics Identifications. Available online: https://bioconductor.org/packages/release/bioc/html/MSnID.html (accessed on 29 November 2019).
- Dickhut, C.; Feldmann, I.; Lambert, J.; Zahedi, R.P. Impact of digestion conditions on phosphoproteomics. J. Proteome Res. 2014, 13, 2761–2770. [Google Scholar] [CrossRef]
- Cohen, S.A.; Michaud, D.P. Synthesis of a fluorescent derivatizing reagent, 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate, and its application for the analysis of hydrolysate amino acids via high-performance liquid chromatography. Anal. Biochem. 1993, 211, 279–287. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genemutation | Protein Name | Function | Variant | SIS Sequence |
|---|---|---|---|---|
| (Found in) | ||||
| SRPX2E234K | Sushi repeat-containing protein SRPX2 | Ligand for the urokinase plasminogen activator surface receptor. Plays a role in angiogenesis. Involved in cellular migration and adhesion. | Canonical | GPEPGSHFPEGEHVIR |
| (WES + RNAseq) | E234K | GPEPGSHFPK | ||
| KS6A5D554N | Ribosomal protein S6 kinase alpha-5, S6K-alpha-5, EC 2.7.11.1 | Ser/Thr kinase required for the mitogen or stress-induced phosphorylation of transcription factors CREB1, ATF1, and the regulation of the transcription factors RELA, STAT3, ETV1/ER81. | Canonical | DLKPENLLFTDENDNLEIK |
| (WES + RNAseq) | D554N | DLKPENLLFTNENDNLEIK | ||
| NRAS * | SYGIPYIETSAK | |||
| KRASG12V | GTPase KRas | Ras proteins bind GDP/GTP and possess intrinsic GTPase activity. Plays an important role in the regulation of cell proliferation. | HRAS * | SFADINLYR |
| (WES + RNAseq) | H/N/KRAS * | LVVVGAGGVGK | ||
| Canonical | SFEDIHHYR | |||
| G12V | LVVVGAVGVGK | |||
| PTBP1K508E | Polypyrimidine tract-binding protein 1 | Plays a role in pre-mRNA splicing and in the regulation of alternative splicing events. | Canonical | DYGNSPLHR |
| (WES + RNAseq) | Canonical | VLFSSNGGVVK | ||
| K508E | VLFSSNGGVVEGFK | |||
| ARL2V141A | ADP-ribosylation factor-like protein 2 | Small GTP-binding protein which cycles between an inactive GDP-bound and an active GTP-bound form | Canonical | EVLELDSIR |
| (WES) | V141A | EALELDSIR | ||
| PPP1R14CT10A | Protein phosphatase 1 regulatory subunit 14C | Inhibitor of the PP1 regulatory subunit PPP1CA. | Canonical | SVATGSSEATGGASGGGAR |
| (WES) | T10A | SVATGSSEAAGGASGGGAR | ||
| HAUS7T244A | HAUS augmin-like complex subunit 7 | Contributes to mitotic spindle assembly, maintenance of centrosome integrity and completion of cytokinesis as part of the HAUS augmin-like complex. | Canonical | TEYFAQHEQGAAAGAADISTLDQK |
| (WES + RNAseq) | T244A | AEYFAQHEQGAAAGAADISTLDQK | ||
| TBD2BA8G | TBC1 domain family member 2B | May act as a GTPase-activating protein. | Canonical | AEEGGGGGEGAAQGAAAEPGAGPAR |
| (WES) | A8G | GEEGGGGGEGAAQGAAAEPGAGPAR |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blank-Landeshammer, B.; Richard, V.R.; Mitsa, G.; Marques, M.; LeBlanc, A.; Kollipara, L.; Feldmann, I.; Couetoux du Tertre, M.; Gambaro, K.; McNamara, S.; et al. Proteogenomics of Colorectal Cancer Liver Metastases: Complementing Precision Oncology with Phenotypic Data. Cancers 2019, 11, 1907. https://doi.org/10.3390/cancers11121907
Blank-Landeshammer B, Richard VR, Mitsa G, Marques M, LeBlanc A, Kollipara L, Feldmann I, Couetoux du Tertre M, Gambaro K, McNamara S, et al. Proteogenomics of Colorectal Cancer Liver Metastases: Complementing Precision Oncology with Phenotypic Data. Cancers. 2019; 11(12):1907. https://doi.org/10.3390/cancers11121907
Chicago/Turabian StyleBlank-Landeshammer, Bernhard, Vincent R. Richard, Georgia Mitsa, Maud Marques, André LeBlanc, Laxmikanth Kollipara, Ingo Feldmann, Mathilde Couetoux du Tertre, Karen Gambaro, Suzan McNamara, and et al. 2019. "Proteogenomics of Colorectal Cancer Liver Metastases: Complementing Precision Oncology with Phenotypic Data" Cancers 11, no. 12: 1907. https://doi.org/10.3390/cancers11121907
APA StyleBlank-Landeshammer, B., Richard, V. R., Mitsa, G., Marques, M., LeBlanc, A., Kollipara, L., Feldmann, I., Couetoux du Tertre, M., Gambaro, K., McNamara, S., Spatz, A., Zahedi, R. P., Sickmann, A., Batist, G., & Borchers, C. H. (2019). Proteogenomics of Colorectal Cancer Liver Metastases: Complementing Precision Oncology with Phenotypic Data. Cancers, 11(12), 1907. https://doi.org/10.3390/cancers11121907