LSD1/KDM1A, a Gate-Keeper of Cancer Stemness and a Promising Therapeutic Target



Abstract
1. Introduction
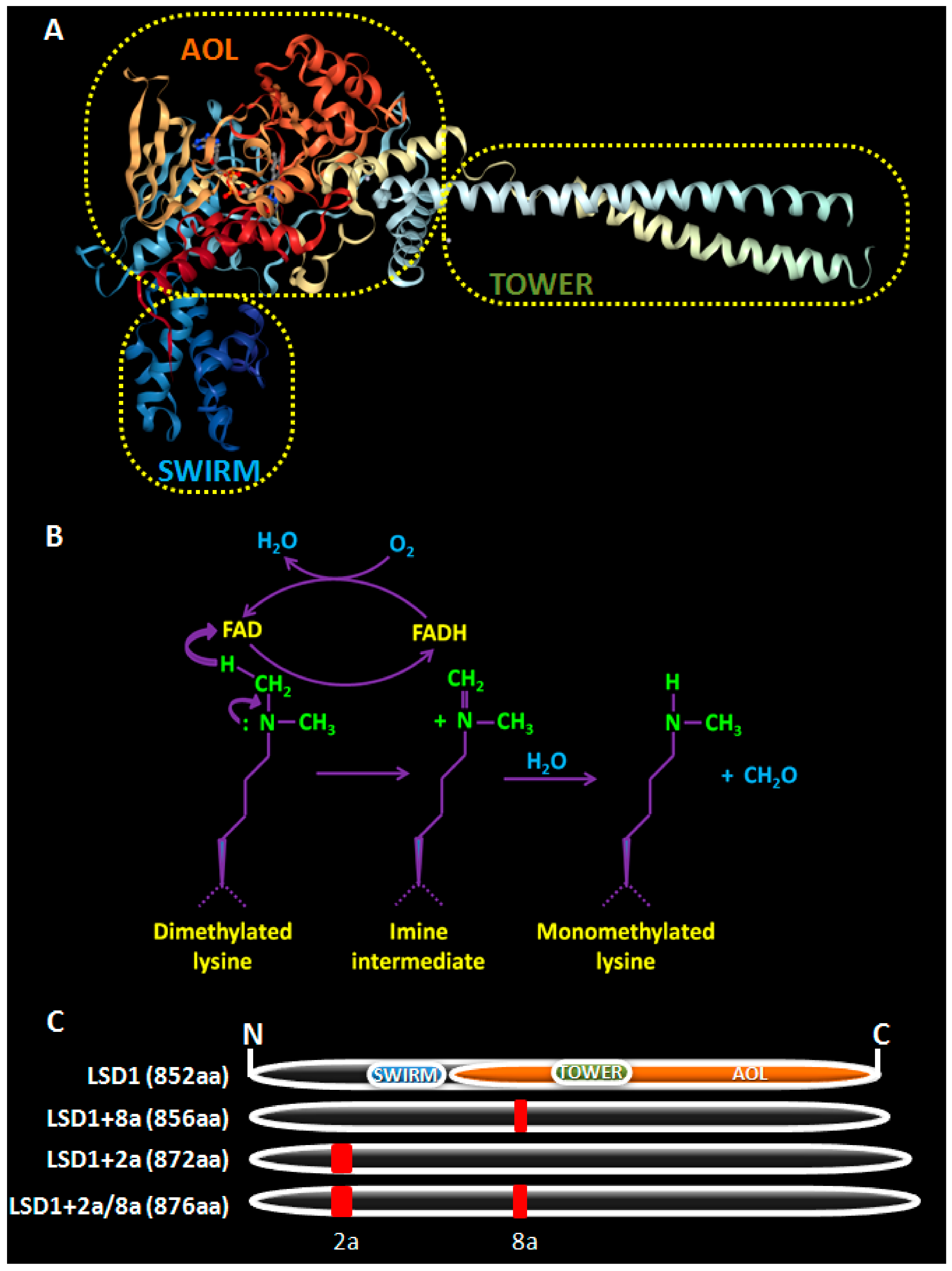
2. LSD1, a Brief Overview
3. LSD1 as a Regulator of Tissue and Cancer Stem Cells
3.1. LSD1 in Tissue Stem Cells
3.1.1. LSD1 in Normal Hematopoietic Stem Cells
3.1.2. LSD1 in Neural Stem/Progenitor Cells (NSPCs)
3.1.3. LSD1 in Other Normal Stem Cell Types
3.2. LSD1 and CSCs
3.2.1. LSD1 in Malignant Hematopoietic Stem Cells
3.2.2. LDS1 in Glioblastoma Stem Cells
3.2.3. LSD1 in Hepatocellular Carcinoma Stem Cells
3.2.4. LSD1 in Breast Cancer Stem Cells
3.2.5. LSD1 in Other Types of CSCs
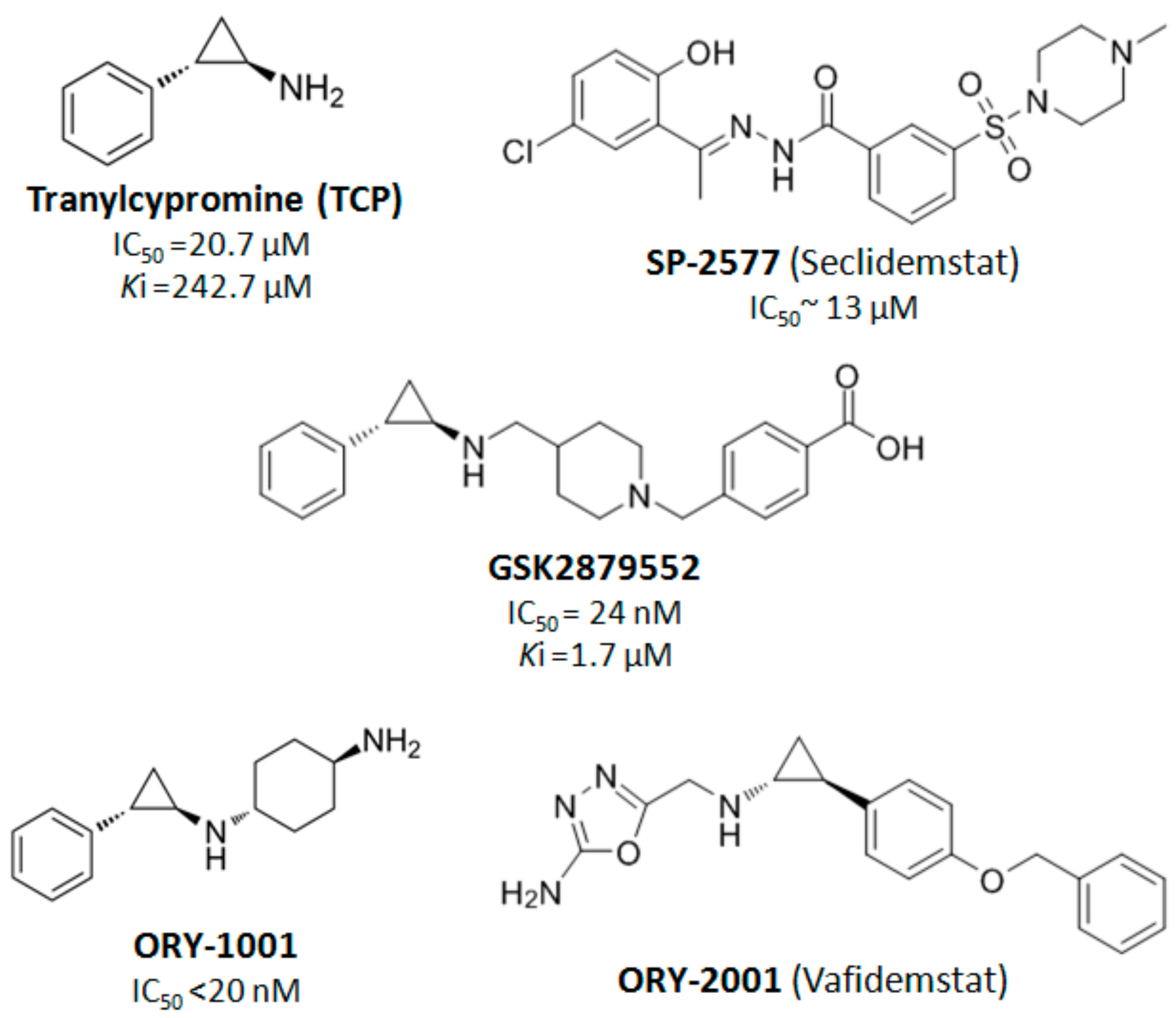
4. LSD1 as a Therapeutic Target in Cancer Treatment
5. Conclusions
Funding
Conflicts of Interest
References
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Højfeldt, J.W.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Thinnes, C.C.; England, K.S.; Kawamura, A.; Chowdhury, R.; Schofield, C.J.; Hopkinson, R.J. Targeting histone lysine demethylases—Progress, challenges, and the future. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2014, 1839, 1416–1432. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, E.; Turberfield, A.H.; Klose, R.J. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015, 16, 1620–1639. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Adamo, A.; Sesé, B.; Boue, S.; Castaño, J.; Paramonov, I.; Barrero, M.J.; Belmonte, J.C.I. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat. Cell Biol. 2011, 13, 652–661. [Google Scholar] [CrossRef]
- Cho, H.S.; Suzuki, T.; Dohmae, N.; Hayami, S.; Unoki, M.; Yoshimatsu, M.; Toyokawa, G.; Takawa, M.; Chen, T.; Kurash, J.K.; et al. Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res. 2011, 71, 655–660. [Google Scholar] [CrossRef]
- Nakao, M.; Anan, K.; Araki, H.; Hino, S. Distinct Roles of the NAD+-Sirt1 and FAD-LSD1 Pathways in Metabolic Response and Tissue Development. Trends Endocrinol. Metab. 2019, 30, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, S.; Saccà, C.D.; Majello, B. Epigenetic regulation of epithelial to mesenchymal transition by the Lysine-specific demethylase LSD1/KDM1A. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2017, 1860, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.J.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.H.; Lim, S.; Schramm, A.; Friedrichs, N.; Koster, J.; Versteeg, R.; Ora, I.; Pajtler, K.; Klein-Hitpass, L.; Kuhfittig-Kulle, S.; et al. Lysine-Specific Demethylase 1 Is Strongly Expressed in Poorly Differentiated Neuroblastoma: Implications for Therapy. Cancer Res. 2009, 69, 2065–2071. [Google Scholar] [CrossRef]
- Theisen, E.R.; Gajiwala, S.; Bearss, J.; Sorna, V.; Sharma, S.; Janat-Amsbury, M. Reversible inhibition of lysine specific demethylase 1 is a novel anti-tumor strategy for poorly differentiated endometrial carcinoma. BMC Cancer 2014, 14, 752. [Google Scholar] [CrossRef]
- Nagasawa, S.; Sedukhina, A.S.; Nakagawa, Y.; Maeda, I.; Kubota, M.; Ohnuma, S.; Tsugawa, K.; Ohta, T.; Roche-Molina, M.; Bernal, J.A.; et al. LSD1 overexpression is associated with poor prognosis in basal-like breast cancer, and sensitivity to PARP inhibition. PLoS ONE 2015, 10, e0118002. [Google Scholar] [CrossRef]
- Martello, G.; Smith, A. The Nature of Embryonic Stem Cells. Annu. Rev. Cell Dev. Biol. 2014, 30, 647–675. [Google Scholar] [CrossRef]
- Raff, M. Adult Stem Cell Plasticity: Fact or Artifact? Annu. Rev. Cell Dev. Biol. 2003, 19, 1–22. [Google Scholar] [CrossRef]
- Bilic, J.; Izpisua Belmonte, J.C. Concise review: Induced pluripotent stem cells versus embryonic stem cells: Close enough or yet too far apart? Stem Cells 2012, 30, 33–41. [Google Scholar] [CrossRef]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Hosseini, A.; Minucci, S. A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics 2017, 9, 1123–1142. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Lania, L.; Majello, B. The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2013, 1829, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Y.; Wang, F.; Wan, K.; Yamane, K.; Zhang, Y.; Lei, M. Crystal structure of human histone lysine-specific demethlase 1 (LSD1). Proc. Natl. Acad. Sci. USA 2006, 103, 13956–13961. [Google Scholar] [CrossRef] [PubMed]
- Da, G.; Lenkart, J.; Zhao, K.; Shiekhattar, R.; Cairns, B.R.; Marmorstein, R. Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc. Natl. Acad. Sci. USA 2006, 103, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.J.; Lei, P.M.; Wong, S.Y.; Ma, D.L.; Leung, C.H. Pharmacological Inhibition of LSD1 for Cancer Treatment. Molecules 2018, 23, 3194. [Google Scholar] [CrossRef] [PubMed]
- Maiques-Diaz, A.; Somervaille, T.C.P. LSD1: Biologic roles and therapeutic targeting. Epigenomics 2016, 8, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.F.M.; Günther, T.; Buettner, R.; Schüle, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Garcia-Bassets, I.; Kwon, Y.S.; Telese, F.; Prefontaine, G.G.; Hutt, K.R.; Cheng, C.S.; Ju, B.G.; Ohgi, K.A.; Wang, J.; Escoubet-Lozach, L.; et al. Histone Methylation-Dependent Mechanisms Impose Ligand Dependency for Gene Activation by Nuclear Receptors. Cell 2007, 128, 505–518. [Google Scholar] [CrossRef]
- Zibetti, C.; Adamo, A.; Binda, C.; Forneris, F.; Toffolo, E.; Verpelli, C.; Ginelli, E.; Mattevi, A.; Sala, C.; Battaglioli, E. Alternative Splicing of the Histone Demethylase LSD1/KDM1 Contributes to the Modulation of Neurite Morphogenesis in the Mammalian Nervous System. J. Neurosci. 2010, 30, 2521–2532. [Google Scholar] [CrossRef]
- Rusconi, F.; Grillo, B.; Toffolo, E.; Mattevi, A.; Battaglioli, E. NeuroLSD1: Splicing-Generated Epigenetic Enhancer of Neuroplasticity. Trends Neurosci. 2017, 40, 28–38. [Google Scholar] [CrossRef]
- Wang, J.; Telese, F.; Tan, Y.; Li, W.; Jin, C.; He, X.; Basnet, H.; Ma, Q.; Merkurjev, D.; Zhu, X.; et al. LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat. Neurosci. 2015, 18, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, P.; Blobel, G.; Hoelz, A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat. Struct. Mol. Biol. 2006, 13, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlic, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef]
- Kontaki, H.; Talianidis, I. Lysine Methylation Regulates E2F1-Induced Cell Death. Mol. Cell 2010, 39, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009, 41, 125–129. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, J.H.; Choi, H.J.; Won, H.Y.; Joo, H.S.; Shin, D.H.; Park, M.K.; Han, B.; Kim, K.P.; Lee, T.J.; et al. LSD1 demethylates HIF1α to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene 2017, 36, 5512–5521. [Google Scholar] [CrossRef]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef]
- Saleque, S.; Kim, J.; Rooke, H.M.; Orkin, S.H. Epigenetic Regulation of Hematopoietic Differentiation by Gfi-1 and Gfi-1b Is Mediated by the Cofactors CoREST and LSD1. Mol. Cell 2007, 27, 562–572. [Google Scholar] [CrossRef]
- Thambyrajah, R.; Mazan, M.; Patel, R.; Moignard, V.; Stefanska, M.; Marinopoulou, E.; Li, Y.; Lancrin, C.; Clapes, T.; Möroÿ, T.; et al. GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell Biol. 2016, 18, 21–32. [Google Scholar] [CrossRef]
- Lancrin, C.; Sroczynska, P.; Stephenson, C.; Allen, T.; Kouskoff, V.; Lacaud, G. The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature 2009, 457, 892–895. [Google Scholar] [CrossRef] [PubMed]
- Sprüssel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Händschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; König, K.; Diehl, L.; et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef] [PubMed]
- Kerenyi, M.A.; Shao, Z.; Hsu, Y.J.; Guo, G.; Luc, S.; O’Brien, K.; Fujiwara, Y.; Peng, C.; Nguyen, M.; Orkin, S.H. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife 2013, 2, e00633. [Google Scholar] [CrossRef] [PubMed]
- Hitoshi, S.; Alexson, T.; Tropepe, V.; Donoviel, D.; Elia, A.J.; Nye, J.S.; Conlon, R.A.; Mak, T.W.; Bernstein, A.; van der Kooy, D. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 2002, 16, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Namihira, M. LSD1 Mediates Neuronal Differentiation of Human Fetal Neural Stem Cells by Controlling the Expression of a Novel Target Gene, HEYL. Stem Cells 2016, 34, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Scully, K.; Zhu, X.; Cai, L.; Zhang, J.; Prefontaine, G.G.; Krones, A.; Ohgi, K.A.; Zhu, P.; Garcia-Bassets, I.; et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 2007, 446, 882–887. [Google Scholar] [CrossRef]
- Lopez, C.I.; Saud, K.E.; Aguilar, R.; Berndt, F.A.; Cánovas, J.; Montecino, M.; Kukuljan, M. The chromatin modifying complex CoREST/LSD1 negatively regulates notch pathway during cerebral cortex development. Dev. Neurobiol. 2016, 76, 1360–1373. [Google Scholar] [CrossRef]
- Fuentes, P.; Cánovas, J.; Berndt, F.A.; Noctor, S.C.; Kukuljan, M. CoREST/LSD1 control the development of pyramidal cortical neurons. Cereb. Cortex 2012, 22, 1431–1441. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Q.; Yang, P.; Wang, C.; Liu, J.; Ding, W.; Liu, W.; Bai, Y.; Yang, Y.; Wang, H.; et al. LSD1 co-repressor Rcor2 orchestrates neurogenesis in the developing mouse brain. Nat. Commun. 2016, 7, 10481. [Google Scholar] [CrossRef]
- Han, X.; Gui, B.; Xiong, C.; Zhao, L.; Liang, J.; Sun, L.; Yang, X.; Yu, W.; Si, W.; Yan, R.; et al. Destabilizing LSD1 by Jade-2 promotes neurogenesis: An antibraking system in neural development. Mol. Cell 2014, 55, 482–494. [Google Scholar] [CrossRef]
- Mahmoudi, E.; Cairns, M.J. MiR-137: An important player in neural development and neoplastic transformation. Mol. Psychiatry 2017, 22, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Ye, P.; Murai, K.; Lang, M.F.; Li, S.; Zhang, H.; Li, W.; Fu, C.; Yin, J.; Wang, A.; et al. MiR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat. Commun. 2011, 2, 510–529. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yu, R.T.; Evans, R.M.; Lie, D.C.; Taupin, P.; Nakashima, K.; Ray, J.; Gage, F.H. Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature 2004, 427, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Alzayady, K.; Stewart, R.; Ye, P.; Yang, S.; Li, W.; Shi, Y. Histone Demethylase LSD1 Regulates Neural Stem Cell Proliferation. Mol. Cell. Biol. 2010, 30, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xu, D.; Yuan, L.; Sun, Y.; Xu, Z. Epigenetic regulation of Atrophin1 by lysine-specific demethylase 1 is required for cortical progenitor maintenance. Nat. Commun. 2014, 5, 5815. [Google Scholar] [CrossRef]
- Patel, D.; Shimomura, A.; Majumdar, S.; Holley, M.C.; Hashino, E. The histone demethylase LSD1 regulates inner ear progenitor differentiation through interactions with Pax2 and the NuRD repressor complex. PLoS ONE 2018, 13, e0191689. [Google Scholar] [CrossRef]
- Popova, E.Y.; Pinzon-Guzman, C.; Salzberg, A.C.; Zhang, S.S.M.; Barnstable, C.J. LSD1-Mediated Demethylation of H3K4me2 Is Required for the Transition from Late Progenitor to Differentiated Mouse Rod Photoreceptor. Mol. Neurobiol. 2016, 53, 4563–4581. [Google Scholar] [CrossRef]
- Krolewski, R.C.; Packard, A.; Schwob, J.E. Global expression profiling of globose basal cells and neurogenic progression within the olfactory epithelium. J. Comp. Neurol. 2013, 521, 833–859. [Google Scholar] [CrossRef]
- Toffolo, E.; Rusconi, F.; Paganini, L.; Tortorici, M.; Pilotto, S.; Heise, C.; Verpelli, C.; Tedeschi, G.; Maffioli, E.; Sala, C.; et al. Phosphorylation of neuronal Lysine-Specific Demethylase 1LSD1/KDM1A impairs transcriptional repression by regulating interaction with CoREST and histone deacetylases HDAC1/2. J. Neurochem. 2014, 128, 603–616. [Google Scholar] [CrossRef]
- Hwang, I.; Cao, D.; Na, Y.; Kim, D.Y.; Zhang, T.; Yao, J.; Oh, H.; Hu, J.; Zheng, H.; Yao, Y.; et al. Far Upstream Element-Binding Protein 1 Regulates LSD1 Alternative Splicing to Promote Terminal Differentiation of Neural Progenitors. Stem Cell Rep. 2018, 10, 1208–1221. [Google Scholar] [CrossRef]
- Laurent, B.; Ruitu, L.; Murn, J.; Hempel, K.; Ferrao, R.; Xiang, Y.; Liu, S.; Garcia, B.A.; Wu, H.; Wu, F.; et al. A Specific LSD1/KDM1A Isoform Regulates Neuronal Differentiation through H3K9 Demethylation. Mol. Cell 2015, 57, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Lambrot, R.; Lafleur, C.; Kimmins, S. The histone demethylase KDM1A is essential for the maintenance and differentiation of spermatogonial stem cells and progenitors. FASEB J. 2015, 29, 4402–4416. [Google Scholar] [CrossRef] [PubMed]
- Myrick, D.A.; Christopher, M.A.; Scott, A.M.; Simon, A.K.; Donlin-Asp, P.G.; Kelly, W.G.; Katz, D.J. KDM1A/LSD1 regulates the differentiation and maintenance of spermatogonia in mice. PLoS ONE 2017, 12, e0177473. [Google Scholar] [CrossRef] [PubMed]
- Musri, M.M.; Carmona, M.C.; Hanzu, F.A.; Kaliman, P.; Gomis, R.; Párrizas, M. Histone demethylase LSD1 regulates adipogenesis. J. Biol. Chem. 2010, 285, 30034–30041. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.J.; Park, U.H.; Kim, J.W.; Choi, H.; Um, S.J.; Kim, E.J. CACUL1 reciprocally regulates SIRT1 and LSD1 to repress PPARγ and inhibit adipogenesis. Cell Death Dis. 2017, 8, 3201. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Liu, Y.; Chen, T.; Zhang, X.; Lv, L.; Jin, C.; Jiang, Y.; Shi, L.; Zhou, Y. The epigenetic promotion of osteogenic differentiation of human adipose-derived stem cells by the genetic and chemical blockade of histone demethylase LSD1. Biomaterials 2014, 35, 6015–6025. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Fan, C.; Wang, Y.J.; Du, Y.G.; Zhu, Y.; Liu, H.; Lv, L.W.; Liu, Y.S.; Zhou, Y.S. MiR-137 knockdown promotes the osteogenic differentiation of human adipose-derived stem cells via the LSD1/BMP2/SMAD4 signaling network. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Lv, L.W.; Ge, W.S.; Liu, Y.S.; Lai, G.Y.; Liu, H.; Li, W.Y.; Zhou, Y.S. Lysine-specific demethylase 1 inhibitor rescues the osteogenic ability of mesenchymal stem cells under osteoporotic conditions by modulating H3K4 methylation. Bone Res. 2016, 4, 16037. [Google Scholar] [CrossRef]
- Sun, J.; Ermann, J.; Niu, N.N.; Yan, G.; Yang, Y.; Shi, Y.J.; Zou, W.G. Histone demethylase LSD1 regulates bone mass by controlling WNT7B and BMP2 signaling in osteoblasts. Bone Res. 2018, 6, 14. [Google Scholar] [CrossRef]
- Adamik, J.; Jin, S.; Sun, Q.; Zhang, P.; Weiss, K.R.; Anderson, J.L.; Silbermann, R.; Roodman, G.D.; Galson, D.L. EZH2 or HDAC1 Inhibition Reverses Multiple Myeloma–Induced Epigenetic Suppression of Osteoblast Differentiation. Mol. Cancer Res. 2017, 15, 405–417. [Google Scholar] [CrossRef]
- Meng, X.; Li, J.; Zheng, M.; Zuo, L.; Sun, C.; Zhu, Y.; Fang, L.; Liu, L.; Zhou, X. Stable H3 peptide was delivered by gold nanorods to inhibit LSD1 activation and induce human mesenchymal stem cells differentiation. Oncotarget 2017, 8, 23110–23119. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tosic, M.; Allen, A.; Willmann, D.; Lepper, C.; Kim, J.; Duteil, D.; Schüle, R. Lsd1 regulates skeletal muscle regeneration and directs the fate of satellite cells. Nat. Commun. 2018, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Scionti, I.; Hayashi, S.; Mouradian, S.; Girard, E.; Esteves de Lima, J.; Morel, V.; Simonet, T.; Wurmser, M.; Maire, P.; Ancelin, K.; et al. LSD1 Controls Timely MyoD Expression via MyoD Core Enhancer Transcription. Cell Rep. 2017, 18, 1996–2006. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.F.; Zhang, J.; Tian, Z.Q.; Wu, Y.Z. Epigenetics in cancer stem cells. Yi Chuan 2013, 35, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer Epigenetics: Tumor Heterogeneity, Plasticity of Stem-like States, and Drug Resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef]
- Hinohara, K.; Polyak, K. Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef]
- Rice, K.L.; Hormaeche, I.; Licht, J.D. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene 2007, 26, 6697–6714. [Google Scholar] [CrossRef]
- Andricovich, J.; Kai, Y.; Tzatsos, A. Lysine-specific histone demethylases in normal and malignant hematopoiesis. Exp. Hematol. 2016, 44, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Goyama, S.; Kitamura, T. Epigenetics in normal and malignant hematopoiesis: An overview and update 2017. Cancer Sci. 2017, 108, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Castelli, G.; Pelosi, E.; Testa, U. Targeting histone methyltransferase and demethylase in acute myeloid leukemia therapy. Onco Targets Ther. 2017, 11, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.J.; Huang, X.; Lynch, J.T.; Spencer, G.J.; Hitchin, J.R.; Li, Y.; Ciceri, F.; Blaser, J.G.; Greystoke, B.F.; Jordan, A.M.; et al. The Histone Demethylase KDM1A Sustains the Oncogenic Potential of MLL-AF9 Leukemia Stem Cells. Cancer Cell 2012, 21, 473–487. [Google Scholar] [CrossRef]
- Cusan, M.; Cai, S.F.; Mohammad, H.P.; Krivtsov, A.; Chramiec, A.; Loizou, E.; Witkin, M.D.; Smitheman, K.N.; Tenen, D.G.; Ye, M.; et al. LSD1 inhibition exerts its antileukemic effect by recommissioning PU.1-and C/EBPα-dependent enhancers in AML. Blood 2018, 131, 1730–1742. [Google Scholar] [CrossRef]
- Schenk, T.; Chen, W.C.; Göllner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef]
- Maiques-Diaz, A.; Spencer, G.J.; Lynch, J.T.; Ciceri, F.; Williams, E.L.; Amaral, F.M.R.; Wiseman, D.H.; Harris, W.J.; Li, Y.; Sahoo, S.; et al. Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep. 2018, 22, 3641–3659. [Google Scholar] [CrossRef]
- Vinyard, M.E.; Su, C.; Siegenfeld, A.P.; Waterbury, A.L.; Freedy, A.M.; Gosavi, P.M.; Park, Y.; Kwan, E.E.; Senzer, B.D.; Doench, J.G.; et al. CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML. Nat. Chem. Biol. 2019, 15, 529–539. [Google Scholar] [CrossRef]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33, 495–511. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Gamo, K.; Yabuki, M.; Takagi, S.; Toyoshima, K.; Nakayama, K.; Nakayama, A.; Morimoto, M.; Miyashita, H.; Dairiki, R.; et al. A Novel LSD1 Inhibitor T-3775440 Disrupts GFI1B-Containing Complex Leading to Transdifferentiation and Impaired Growth of AML Cells. Mol. Cancer Ther. 2016, 16, 273–284. [Google Scholar] [CrossRef]
- Wada, T.; Koyama, D.; Kikuchi, J.; Honda, H.; Furukawa, Y. Overexpression of the shortest isoform of histone demethylase LSD1 primes hematopoietic stem cells for malignant transformation. Blood 2015, 125, 3731–3745. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.; Takouda, J.; Yoshimoto, K.; Nakashima, K. New aspects of glioblastoma multiforme revealed by similarities between neural and glioblastoma stem cells. Cell Biol. Toxicol. 2018, 34, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Matarredona, E.R.; Pastor, A.M. Neural Stem Cells of the Subventricular Zone as the Origin of Human Glioblastoma Stem Cells. Therapeutic Implications. Front. Oncol. 2019, 9, 779. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Viswanadhapalli, S.; Surapaneni, P.; Suzuki, T.; Brenner, A.; Vadlamudi, R.K. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene 2017, 36, 2423–2434. [Google Scholar] [CrossRef]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; DePinho, R.A.; et al. Olig2-Regulated Lineage-Restricted Pathway Controls Replication Competence in Neural Stem Cells and Malignant Glioma. Neuron 2007, 53, 503–517. [Google Scholar] [CrossRef]
- Piccirillo, S.G.M.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar] [CrossRef]
- Zhou, A.; Lin, K.; Zhang, S.; Chen, Y.; Zhang, N.; Xue, J.; Wang, Z.; Aldape, K.D.; Xie, K.; Woodgett, J.R.; et al. Nuclear GSK3β promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat. Cell Biol. 2016, 18, 954–966. [Google Scholar] [CrossRef]
- Hiramatsu, H.; Kobayashi, K.; Kobayashi, K.; Haraguchi, T.; Ino, Y.; Todo, T.; Iba, H. The role of the SWI/SNF chromatin remodeling complex in maintaining the stemness of glioma initiating cells. Sci. Rep. 2017, 7, 889. [Google Scholar] [CrossRef]
- Lei, Z.J.; Wang, J.; Xiao, H.L.; Guo, Y.; Wang, T.; Li, Q.; Liu, L.; Luo, X.; Fan, L.L.; Lin, L.; et al. Lysine-specific demethylase 1 promotes the stemness and chemoresistance of Lgr5+ liver cancer initiating cells by suppressing negative regulators of β-catenin signaling. Oncogene 2015, 34, 3188–3198. [Google Scholar] [CrossRef]
- Huang, M.; Chen, C.; Geng, J.; Han, D.; Wang, T.; Xie, T.; Wang, L.; Wang, Y.; Wang, C.; Lei, Z.; et al. Targeting KDM1A attenuates Wnt/β-catenin signaling pathway to eliminate sorafenib-resistant stem-like cells in hepatocellular carcinoma. Cancer Lett. 2017, 398, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, L.; Chen, X.; Cheng, J.; Zhang, H.; Zhang, C.; Shan, J.; Shen, J.; Qian, C. LSD1 stimulates cancer-Associated fibroblasts to drive Notch3-Dependent self-Renewal of liver cancer stem–like cells. Cancer Res. 2018, 78, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Yang, X.H.; Kang, T.; Zhao, Y.; Wang, C.; Evers, B.M.; Zhou, B.P. The Deubiquitinase USP28 Stabilizes LSD1 and Confers Stem-Cell-like Traits to Breast Cancer Cells. Cell Rep. 2013, 5, 224–236. [Google Scholar] [CrossRef]
- Boulding, T.; McCuaig, R.D.; Tan, A.; Hardy, K.; Wu, F.; Dunn, J.; Kalimutho, M.; Sutton, C.R.; Forwood, J.K.; Bert, A.G.; et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci. Rep. 2018, 8, 73. [Google Scholar] [CrossRef]
- Verigos, J.; Karakaidos, P.; Kordias, D.; Papoudou-Bai, A.; Evangelou, Z.; Harissis, H.V.; Klinakis, A.; Magklara, A. The Histone Demethylase LSD1/ΚDM1A Mediates Chemoresistance in Breast Cancer via Regulation of a Stem Cell Program. Cancers 2019, 11, 1585. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. The SNAG domain of snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Li, X.Y.; Hu, C.Y.; Ford, M.; Kleer, C.G.; Weiss, S.J. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. USA 2012, 109, 16654–16659. [Google Scholar] [CrossRef]
- Ferrari-Amorotti, G.; Chiodoni, C.; Shen, F.; Cattelani, S.; Soliera, A.R.; Manzotti, G.; Grisendi, G.; Dominici, M.; Rivasi, F.; Colombo, M.P.; et al. Suppression of Invasion and Metastasis of Triple-Negative Breast Cancer Lines by Pharmacological or Genetic Inhibition of Slug Activity. Neoplasia 2014, 16, 1047–1058. [Google Scholar] [CrossRef]
- Feng, J.; Xu, G.; Liu, J.; Zhang, N.; Li, L.; Ji, J.; Zhang, J.; Zhang, L.; Wang, G.; Wang, X.; et al. Phosphorylation of LSD1 at Ser112 is crucial for its function in induction of EMT and metastasis in breast cancer. Breast Cancer Res. Treat. 2016, 159, 443–456. [Google Scholar] [CrossRef]
- Choi, H.J.; Park, J.H.; Park, M.; Won, H.Y.; Joo, H.S.; Lee, C.H.; Lee, J.Y.; Kong, G. UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO Rep. 2015, 16, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.C.; Liu, Y.S.; Tseng, K.C.; Yang, T.S.; Yeh, C.Y.; You, J.F.; Hung, H.Y.; Chen, S.J.; Chen, H.C. CBB1003, a lysine-specific demethylase 1 inhibitor, suppresses colorectal cancer cells growth through down-regulation of leucine-rich repeat-containing G-protein-coupled receptor 5 expression. J. Cancer Res. Clin. Oncol. 2015, 141, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, F.; Ren, Q.; Sun, H.; Xu, Z.; Lan, R.; Liu, Y.; Ward, D.; Quan, J.; Ye, T.; et al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011, 71, 7238–7249. [Google Scholar] [CrossRef] [PubMed]
- Hoang, N.; Zhang, X.; Zhang, C.; Vo, V.; Leng, F.; Saxena, L.; Yin, F.; Lu, F.; Zheng, G.; Bhowmik, P.; et al. New histone demethylase LSD1 inhibitor selectively targets teratocarcinoma and embryonic carcinoma cells. Bioorg. Med. Chem. 2018, 26, 1523–1537. [Google Scholar] [CrossRef]
- Wang, D.; Li, W.; Zhao, R.; Chen, L.; Liu, N.; Tian, Y.; Zhao, H.; Xie, M.; Lu, F.; Fang, Q.; et al. Stabilized peptide HDAC inhibitors derived from HDAC1 substrate H3K56 for the treatment of cancer stem–like cells in vivo. Cancer Res. 2019, 79, 1769–1783. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, F.; Wang, J.; Yin, F.; Xu, Z.; Qi, D.; Wu, X.; Cao, Y.; Liang, W.; Liu, Y.; et al. Pluripotent Stem Cell Protein Sox2 Confers Sensitivity to LSD1 Inhibition in Cancer Cells. Cell Rep. 2013, 5, 445–457. [Google Scholar] [CrossRef]
- Romo-Morales, A.; Aladowicz, E.; Blagg, J.; Gatz, S.A.; Shipley, J.M. Catalytic inhibition of KDM1A in Ewing sarcoma is insufficient as a therapeutic strategy. Pediatr. Blood Cancer 2019. [Google Scholar] [CrossRef]
- Sonnemann, J.; Zimmermann, M.; Marx, C.; Ebert, F.; Becker, S.; Lauterjung, M.L.; Beck, J.F. LSD1 (KDM1A)-independent effects of the LSD1 inhibitor SP2509 in cancer cells. Br. J. Haematol. 2018, 183, 494–497. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357. [Google Scholar] [CrossRef]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Xu, S.; Wu, L.; Ma, T.; Liu, R.; Liu, Y.C.; Deng, D.; Gu, Y.; Zhou, J.; Lan, F.; et al. Design, synthesis and biological activity of 3-oxoamino-benzenesulfonamides as selective and reversible LSD1 inhibitors. Bioorg. Chem. 2017, 72, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Stazi, G.; Zwergel, C.; Valente, S.; Mai, A. LSD1 inhibitors: A patent review (2010–2015). Expert Opin. Ther. Pat. 2016, 26, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.M.Z.; McCafferty, D.G. trans-2-phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; VanAller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef]
- Bailey, C.; Romero, M.; Han, R.; Larson, J.; Becher, O.; Lee, D.; Monje, M.; Gopalakrishnan, V.; Zaky, W.; Chandra, J. Immu-19. LSD1 modulates nk cell immunotherapy through an onco-immunogenic gene signature in dipg. Neuro-Oncology 2018, 20 (Suppl. S2), 102. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search (accessed on 6 September 2019).
- World Health Organization, International Clinical Trials Registry Platform. Available online: http://apps.who.int/trialsearch/Default.aspx (accessed on 6 September 2019).
- EU Clinical Trials Register. Available online: https://clinicaltrials.gov/ct2/home (accessed on 6 September 2019).
- Singh, M.M.; Johnson, B.; Venkatarayan, A.; Flores, E.R.; Zhang, J.; Su, X.; Barton, M.; Lang, F.; Chandra, J. Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma. Neuro-Oncology 2015, 17, 1463–1473. [Google Scholar] [CrossRef]
- Magliulo, D.; Bernardi, R.; Messina, S. Lysine-Specific Demethylase 1A as a Promising Target in Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 255. [Google Scholar] [CrossRef]
- Ismail, T.; Lee, H.K.; Kim, C.; Kwon, T.; Park, T.J.; Lee, H.S. KDM1A microenvironment, its oncogenic potential, and therapeutic significance. Epigenet. Chromatin 2018, 11, 33. [Google Scholar] [CrossRef]
- Stazi, G.; Fioravanti, R.; Mai, A.; Mattevi, A.; Valente, S. Histone deacetylases as an epigenetic pillar for the development of hybrid inhibitors in cancer. Curr. Opin. Chem. Biol. 2019, 50, 89–100. [Google Scholar] [CrossRef]
- Duan, Y.C.; Ma, Y.C.; Qin, W.P.; Ding, L.N.; Zheng, Y.C.; Zhu, Y.L.; Zhai, X.Y.; Yang, J.; Ma, C.Y.; Guan, Y.Y. Design and synthesis of tranylcypromine derivatives as novel LSD1/HDACs dual inhibitors for cancer treatment. Eur. J. Med. Chem. 2017, 140, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 2018, 9, 53. [Google Scholar] [CrossRef]
- Anastas, J.N.; Zee, B.M.; Kalin, J.H.; Kim, M.; Guo, R.; Alexandrescu, S.; Blanco, M.A.; Giera, S.; Gillespie, S.M.; Das, J.; et al. Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 2019, 36, 528–544. [Google Scholar] [CrossRef] [PubMed]
- MacK, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stütz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Code | Title | Status | Conditions | Interventions |
|---|---|---|---|---|
| DRKS00006055 | Phase I/II study of sensitization of non-M3 acute myeloid leukemia (AML) blasts to all-trans retinoic acid (ATRA) by epigenetic treatment with tranylcypromine (TCP), an inhibitor of the histone lysine demethylase 1 (LSD1) | Recruiting | AML and Myelodysplastic syndrome | TCP| ATRA| AraC |
| EUCTR2013-002447-29 | A phase I study of Human Pharmacokinetics and Safety of ORY-1001, and LSD1 inhibitor, in relapsed or refractory acute leukaemia (AL) | Not Recruiting | Refractory or Relapsed acute leukaemia | ORY-1001 |
| EUCTR2018-000482-36 | A pilot study to assess the safety, tolerability, dose finding and efficacy of ORY-1001 in combination with azacitidine in older patients with AML in first line therapy | Ongoing | AML | ORY-1001|Etoposide| Carboplatin| Cisplatin |
| NCT02717884 | Study of Sensitization of Non-M3 AML Blasts to ATRA by Epigenetic Treatment With Tranylcypromine (TCP) | Recruiting | AML and Myelodysplastic Syndrome | TCP| ATRA| cytarabine |
| NCT02842827 | IMG-7289, With and Without ATRA, in Patients With Advanced Myeloid Malignancies | Completed | AML and Myelodysplastic Syndrome | IMG-7289| ATRA |
| NCT03136185 | IMG-7289 in Patients With Myelofibrosis | Recruiting | Myelofibrosis| PPV-MF| PET-MF| PMF | IMG-7289 |
| Code | Title | Status | Conditions | Interventions |
|---|---|---|---|---|
| NCT03514407 | A Study of INCB059872 in Relapsed or Refractory Ewing Sarcoma | Recruiting | Relapsed Ewing Sarcoma | INCB059872 |
| NCT03600649 | Clinical Trial of SP-2577 (Seclidemstat) in Patients With Relapsed or Refractory Ewing Sarcoma | Recruiting | Ewing Sarcoma | SP-2577 |
| EUCTR2018-000469-35 | A pilot study to assess the safety, tolerability, dose finding and efficacy ORY-1001 in combination with platinum-etoposide chemotherapy in patients with relapsed, extensive-stage disease small cell lung cancer | Ongoing | Relapsed, extended-stage disease small cell lung cancer | ORY-1001 |
| EUCTR2017-002838-23 | Randomized, double-blind, placebo-controlled, 3-arm, 36 weeks parallel-group study to evaluate the safety and tolerability of ORY-2001 in patients with Relapsing-Remitting Multiple Sclerosis and Secondary Progressive Multiple Sclerosis | Authorised | Relapsing-Remitting Multiple Sclerosis and Secondary Progressive Multiple Sclerosis | ORY2001 |
| NCT03505528 | An Early Phase Study of Abraxane Combined With Phenelzine Sulfate in Patients With Metastatic or Advanced Breast Cancer | Recruiting | Metastatic Breast Cancer | Nanoparticle albumin-bound paclitaxel| Phenelzine Sulfate |
| NCT02712905 | An Open-Label, Dose-Escalation/Dose-Expansion Safety Study of INCB059872 in Subjects With Advanced Malignancies | Recruiting | Solid Tumors and Hematologic Malignancy | INCB059872| ATRA| azacitidine| nivolumab |
| NCT03895684 | Phase 1 Trial of the LSD1 Inhibitor SP-2577 (Seclidemstat) in Patients With Advanced Solid Tumors | Not yet recruiting | Advanced Solid Tumors | SP-2577 Seclidemstat |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karakaidos, P.; Verigos, J.; Magklara, A. LSD1/KDM1A, a Gate-Keeper of Cancer Stemness and a Promising Therapeutic Target. Cancers 2019, 11, 1821. https://doi.org/10.3390/cancers11121821
Karakaidos P, Verigos J, Magklara A. LSD1/KDM1A, a Gate-Keeper of Cancer Stemness and a Promising Therapeutic Target. Cancers. 2019; 11(12):1821. https://doi.org/10.3390/cancers11121821
Chicago/Turabian StyleKarakaidos, Panagiotis, John Verigos, and Angeliki Magklara. 2019. "LSD1/KDM1A, a Gate-Keeper of Cancer Stemness and a Promising Therapeutic Target" Cancers 11, no. 12: 1821. https://doi.org/10.3390/cancers11121821
APA StyleKarakaidos, P., Verigos, J., & Magklara, A. (2019). LSD1/KDM1A, a Gate-Keeper of Cancer Stemness and a Promising Therapeutic Target. Cancers, 11(12), 1821. https://doi.org/10.3390/cancers11121821